
Abstract
Purpose of Review
Traumatic brain injury (TBI) is a leading cause of morbidity and mortality; however, little definitive evidence exists about most clinical management strategies. Here, we highlight important differences between two major guidelines, the 2016 Brain Trauma Foundation guidelines and the Lund Concept, along with recent preclinical and clinical data.
Recent Findings
While intracranial pressure (ICP) monitoring has been questioned, the majority of literature demonstrates benefit in severe TBI. The optimal cerebral perfusion pressure and ICP are yet unknown, but likely as important is the concept of ICP burden. The evidence for antihypertensive therapy is strengthening. Decompressive craniectomy improves mortality, but at the cost of increased morbidity. Plasma-based resuscitation has demonstrated benefit in multiple preclinical TBI studies.
Summary
The management of hemodynamics and intravascular volume are crucial in TBI. Based on recent evidence, ICP monitoring, antihypertensive therapy, minimal use of vasopressors/inotropes, and plasma resuscitation may improve outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
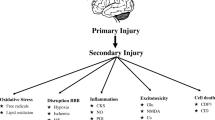
Traumatic brain injury (TBI) is a leading cause of morbidity and mortality in the United States (US) and worldwide. There are over 2 million cases of TBI each year in the US, resulting in approximately 50,000 deaths [1, 2]. There are two main phases of TBI. The first is the primary injury itself, which involves the initial mechanical force of impact, resulting in loss of tissue and neuronal cell death. This primary injury cannot be repaired, and the damage done is permanent. The second phase is related to a hyperexcitatory and inflammatory-mediated secondary injury that occurs during the period of hours to days after the initial injury [3]. This results in increased blood–brain barrier (BBB) permeability and cerebral edema, leading to increasing intracranial pressure, potentially exacerbating cerebral ischemia [4]. The penumbra, the area of the brain surrounding the primary injury, is especially vulnerable to hypoxic insults, and preservation of this region is one of the primary goals in the management of TBI.
The prevention of the secondary brain injury has been the subject of significant research over last 3 decades. However, despite promising data from numerous preclinical trials, no therapy to date has improved outcomes in humans [5]. Furthermore, we still have little definitive evidence about the effectiveness of nearly every aspect of our management of TBI [6]. The guidelines that do exist are based largely on expert consensus and theoretical physiological models based upon animal models. There is no “best’ guideline for treating TBI. The heterogeneity of injury patterns and an initial classification scheme that has numerous potential confounders [i.e., Glasgow Coma Scale (GCS)] hamper the development of effective clinical trials. Further, there may be aspects of TBI, such as sex differences, that we do not yet understand and have yet to adopt in our individual management of TBI [6, 7].
Yet, until a proven therapy to help prevent secondary brain injury is available, we must continue to treat patients suffering TBI in the most empirically effective manner possible. Here, we will highlight fundamental differences between two of the main guidelines available for the management of severe TBI: the most recent Brain Trauma Foundation (BTF) guidelines, published in 2016, and the guidelines prescribed in the Lund Concept (LC) [8, 9••].
Controversies and challenges exist in the process of treating a patient with TBI, from pre-hospital management to long-term, outpatient care [10]. This review, however, will focus on resuscitation during the hours and days after injury, after the patient has been admitted, triaged, and any immediate life-threatening space occupying lesions have been evacuated. Specifically, we will focus on the key physiologic principles that are used in various approaches to minimize secondary brain injury.
Perfusion pressure/hemodynamic strategies in TBI
The intracranial contents include the brain, cerebrospinal fluid, and blood. Intracranial volume is kept constant by the rigid skull; thus, intracranial pressure (ICP) is the sum of its three components. An increase in one component affects the entire compartment. Thus, the brain is rapidly subject to the development of a “compartment syndrome.” Under normal circumstances, ICP is kept relatively constant by compensatory mechanisms, such as autoregulation of cerebral blood flow (CBF) [11]. However, elevations in ICP after TBI—via space occupying lesions, cerebral edema, and disrupted autoregulation—can lead to compression of brain tissue and decrease in CBF, resulting in further ischemic insults and cell death within the brain. Furthermore, MAP changes can have effects on cerebral perfusion pressure (CPP) that would not affect the brain with intact autoregulation [12]. Therefore, each variable in the equation MAP − ICP = CPP must be carefully considered in management to ensure adequate CBF and prevent pathologic elevations in ICP.
CPP and ICP
The parameters for optimal CPP and ICP are not known and are the scrutiny of much research and debate. Further, CPP is not a truly measured variable and is a theoretical entity. There has been a view that ICP elevation is merely an epi-phenomenon that serves as an index of injury severity, but that it is not really a valid therapeutic target. In support of that view, there have been a few studies that claim no difference in outcome with/without ICP monitoring [13]. However, the overwhelming evidence is that ICP/CPP management strategies can be useful in severe TBI [14,15,16]. The critical element distinguishing the two positions is the concept of ICP burden, as this represents the total time spent with an ICP over 25 mm Hg for adults and 20 mm Hg in pediatric patients. Guiza et al., clearly demonstrated the strong association between ICP burden and ultimate outcome using continuous waveform analysis and data capture (see Fig. 1) [17•]. Functionally, all but the shortest interval with an ICP over 20 mm Hg in children is associated with a bad outcome. More than approximately 10 min with an ICP over 25 mm Hg is at high risk for a bad outcome in adults. While there can be disagreement about the optimal strategies to manage this derangement, it is apparent that a pathological ICP is associated with a poor outcome [17•].

Visualization of correlation between Glasgow Outcome Score (GOS) and average number of ICP insults per GOS category. Left adult cohort (n = 261). Right pediatric cohort (n = 99). Each color-coded point in the graph refers to a number of episodes of ICP, defined by a certain ICP intensity threshold (X-axis), and a certain duration threshold (Y-axis). Such an episode is called an ICP insult. The univariate correlation of each type of ICP insult (characterized by ICP intensity and duration thresholds) with outcome is color-coded. Dark red episodes mean that such ICP insults, on average, are associated with worse outcome (lower GOS categories); dark blue episodes mean that such ICP insults, on average, are associated with better outcome (higher GOS categories). The contour of zero correlation is highlighted in black, and is called the transition curve. Reproduced from Ref. [17•], with permission
In contrast to a CPP driven strategy, the LC concept focuses on ICP. The LC attempts to maintain ICP < 20 mm Hg and CPP between 50 and 70 mm Hg, with the caveat that lower CPPs are only tolerated in a normovolemic, non-anemic patient and without the use of beta-agonist inotropic support [18]. The BTF guidelines have adapted their pressure targets in the most recent edition. The threshold at which ICP management should be initiated was increased to 22 mm Hg from 20 mm Hg, while the range of acceptable CPP values was narrowed to 60–70 mm Hg from 50 to 70 mm Hg [8].
The CPP-based strategy is based upon the concept that CPP is a surrogate for CBF, which is highly dependent on cerebral autoregulation. Recent research has highlighted that the degree of disrupted cerebral autoregulation is variable among patients, which can have an effect on their individual optimal CPP [19]. New methods for assessing the autoregulatory capacity of patients have been recently developed; however, they are largely not validated and require sophisticated technologies not present in most centers caring for TBI patients [11, 20, 21]. The fundamental issue is that global pressure perfusion relationships may not be indicative of penumbral regions of disturbed flow or metabolism.
More research is certainly necessary to determine optimal thresholds for ICP and CPP, especially concerning the individual patient’s capacity for cerebral autoregulation. What is evident is that elevations in ICP > 25 mm Hg and deviations in CPP < 50 mm Hg and > 70 mm Hg are likely contribute to secondary brain injury.
Blood Pressure
In TBI, hypotension (defined as SBP < 90 mm Hg) is known to contribute to mortality and worse outcomes in children and adults [22, 23]. Many TBI patients endure simultaneous polytrauma, leading to significant extracranial hemorrhage. Over the last two decades, significant advances in trauma resuscitation have led to improvements in the management of patients with severe hemorrhage; however, morbidity and mortality of patients with TBI and hemorrhagic shock remains high [24, 25].
Furthermore, after the initial resuscitation, the optimal management of hemodynamics in TBI is still under investigation. What is our goal? Is it to lower ICP, with acceptance of lower CPP values? Or do we augment MAP as the primary method of achieving the CPP we desire?
The BTF guidelines recommend maintaining SBP at ≥ 100 mm Hg for patients 50 to 69 year old or at ≥ 110 mm Hg or above for patient 15 to 49 or > 70 years old [8]. Yet, there is only Level III evidence for this recommendation. Furthermore, there is no recommendation for the acceptable upper limit of SBP or management of hypertension. In high-income countries, as morbidity due to cardiovascular disease and cancer has decreased, the incidence of TBI among elderly has increased, with falls being the most common cause of TBI [6]. These patients often have pre-existing hypertension, among other comorbidities, and recent evidence has demonstrated that hypertension may be associated with increased mortality after TBI [26]. Furthermore, catecholamine excess after TBI can lead to elevations in SBP after TBI [9, 26]. In the injured brain, autoregulation mechanism are often impaired, especially at and around the site of injury, and recent evidence has demonstrated that increased SBP and CPP may be directly transmitted to cerebral capillaries, leading to increases in capillary hydrostatic pressure and worsening cerebral edema [9••, 11].
The BTF guidelines provide recommendations for minimum SBP, while hypertension is not specifically addressed. There are no recommendations for or against the use of antihypertensive medications.
Catecholamines and Paroxysmal Sympathetic Hyperactivity After TBI
TBI is known to cause to a catecholamine surge that can result in further damage within the CNS and can have significant effects on the cardiovascular system. The association between brain injury, catecholamines, and cardiac function has been known for some time. In 1971, Hawkins and Clower demonstrated that head trauma in mice caused myocardial damage and necrosis, which was abrogated by administration of reserpine (a sympathetic blocking agent) [27]. Clifton et al. found that the severity of head injury correlated closely with levels of circulating norepinephrine (NE) and dopamine beta-hydroxylase; furthermore, measures of sympathetic activity—including blood pressure, heart rate, and temperature—were similarly elevated in TBI patients with elevated levels of NE [28]. In addition, catecholamine levels were found to correlate with prognosis of patients with Glasgow Coma Scale (GCS) scores of 3 or 4 on admission [29]. This finding from over 30 years ago was confirmed in a recent prospective study, which found that elevated circulating catecholamines were independently associated with functional outcome and mortality in moderate-to-severe TBI [30].
Neuronal injury to specific areas of the brain—such as the insular cortex and subcortical regions—has been implicated in catecholamine release, autonomic dysfunction, and neuroinflammation [31]. The constellation of these pathological alterations has been termed paroxysmal sympathetic hyperactivity (PSH), as there is lack of evidence of specific alterations in parasympathetic activity after neurological injury [32]. Catecholamine excess can lead to mitochondrial dysfunction and cell death within the myocardium. Di Battista et al. demonstrated that levels of catecholamines correlated with circulating cytokine levels, including IL-10, IL-1β, and TNF-α, over the first 24 h after injury. Furthermore, higher levels of systemic inflammatory response correlated with worsened 6-month outcomes [33].
Recent studies have demonstrated that systolic dysfunction is common after TBI, which can further exacerbate secondary brain injury via decreased capacity to maintain cerebral perfusion. Chaikittisilpa et al. found that the presence of systemic inflammatory response syndrome (SIRS) criteria was associated with systolic cardiac dysfunction after TBI [34]. These findings support the notion that catecholamine and systemic inflammatory excess may contribute to worsened outcomes in TBI. Krishnamoorthy et al. have provided evidence that a distinctive hemodynamic profile after TBI, specifically early hypertension and tachycardia, may be predictive of future cardiac dysfunction [35]. It is clear that TBI causes a significant sympathetic nervous system response, including a catecholamine and systemic inflammatory surge, that can result in cardiac dysfunction and damage and correlates with severity of neuronal injury.
Antihypertensive Therapy
From its inception, the LC avoided the use of vasopressors, inotropes, and fluids to maintain supranormal CPP based on physiologic aspects of brain volume regulation in TBI (see Fig. 2; for review of brain volume regulation and the LC, see Grände 2006) [9••, 36, 37]. Instead, the LC advocates to start antihypertensive therapy as soon as the patient has been stabilized. Antihypertensive therapy is used to reduce arterial pressure, cerebral hydrostatic capillary pressure, and adrenergic stress, with avoidance of drugs that induce cerebral vasodilation [9••, 18]. While most TBI guidelines advocate for treating elevations in ICP based on a, incident-driven approach [38, 39], LC advocates maintain that starting antihypertensive therapy early is crucial to preventing pathologic ICP; furthermore, once brain edema develops, reduction is a slow process due to BBB disruption in the brain [9••]. In the LC, beta-1 blockade and alpha-2 agonists are the initial therapies of choice. The principles of hemodynamic management in the LC are targeted towards normotension decreasing catecholamine-induced stress in order to reduce cerebral edema and ICP. However, there is no set definition of normotension or management of SBP with respect to age or comorbidities.

a A schematic illustration of the cerebral capillary and the forces responsible for transcapillary fluid exchange in the uninjured brain with intact BBB. b The cerebral capillary and forces responsible for transcapillary fluid exchange in the injured brain, in which the capillaries are passively permeable for small solutes. Reproduced from Ref. [37], with permission
Beta-Blockers
A great deal of preclinical and clinical research has investigated the mechanisms and effect of beta-blockade in TBI, with promising results. Although not specifically recommended in the BTF guidelines, beta-blockers have become increasingly utilized in the treatment of TBI. The Eastern Association for the Surgery of Trauma recently released guidelines, based on systematic review and meta-analysis, recommending the use of beta-blockers in severe TBI in patients under specific conditions: (1) those treated in an intensive care unit (ICU) where adverse cardiovascular events can be monitored and (2) only if symptomatic hypotension and bradycardia are avoided. Although the guidelines note that while the beneficial effects of beta-blockade in TBI are consistent across multiple studies, prospective randomized trials are still needed [40].
As mentioned above, the beneficial effects of sympathetic nervous system blockade after brain injury have been studied and known for many decades. Hawkins demonstrated that TBI can cause myocardial damage, which was significantly reduced when mice were pretreated with reserpine, which blocks the release of catecholamines [27]. In a mouse model, propranolol, a non-selective beta-blocker, administration at 15 min or 60 min after injury increased cerebral perfusion in a dose-dependent manner [41]. Early propranolol administration has also been shown to attenuate the reduction in cerebral glucose metabolism often seen after TBI and improve motor function testing at 24 h after injury in mice [42]. Beta-adrenergic receptor knockout mice were reported to have decreased fibrinolysis compared to wild-type mice with TBI, potentially leading to attenuation of coagulopathy after TBI and reduction of intracranial hemorrhage progression [43]. While not a TBI-specific model, surgically induced traumatic stress was found to increase levels of pro-inflammatory cytokines in microglia cells, central immune effector cells in the CNS; beta-blockade with propranolol immediately prior to surgery was found to decrease the pro-inflammatory response in isolated microglia [44]. In studying the effects of propranolol and mesenchymal stem cell therapy in TBI, Kota et al. demonstrated that a single dose of propranolol reduced cerebral edema and altered microglia activation [45]. However, there are also data that suggest propranolol can actually increase levels of epinephrine after TBI [46]. Further information is needed to elucidate the mechanism of beta-adrenergic receptors and beta-blockade therapy in TBI.
The beneficial effects of beta-blockade after TBI are also demonstrated across multiple clinical studies [47,48,49,50,51,52]. In a prospective, observational study, Ahl et al. found that early initiation of beta-blocker therapy with continuous exposure during the hospital course decreased length of stay (LOS) and improved functional outcomes at long-term follow-up, although there was no difference in mortality [52]. Another prospective, observational study of early and continuous beta-blocker therapy also demonstrated that beta-blockade was associated with decrease in ICU and hospital LOS without increase risk of bradycardic or hypotensive events [53]. In a prospective, randomized study of severely injured trauma patients, early treatment with propranolol was found to improve bone marrow dysfunction, blunt early tachycardia, and demonstrated a trend towards faster resolution of anemia [54]. Clearly, a prospective randomized trial is needed to assess the efficacy and impact of beta-blocker therapy in TBI. The results from two ongoing randomized clinical trials will hopefully help guide future therapy (NCT02957331 and NTC01322048).
Alpha-2 Agonists
Alpha-2 agonists reduce blood pressure via antisympathetic and sedative effects, and dampen the hyperadrenergic state after TBI [9••, 55]. While non-selective alpha agonists can have vasoconstrictor effects via alpha-1 stimulation, more selective alpha-2 agonist (such as dexmedetomidine) can avoid this and are recommended in low dose by the LC. The critical element is that these drugs work at the pre-capillary level, thus reducing transcapillary hydrostatic pressure and the potential for worsening cerebral edema. While not as extensively studied as beta-blockers in TBI, proponents of the LC point to multiple preclinical TBI studies that showed neuroprotective effects and decreases in plasma catecholamine levels with the use of alpha-2 agonists [56, 57]. Furthermore, there is recent clinical evidence that dexmedetomidine is safe in patients with severe TBI and may decrease requirements for narcotics and additional sedatives [58].
Angiotensin II Antagonists
If beta-blockade and alpha-2 agonists cannot confer normotension, angiotensin II blockade is added as an additional antihypertensive agent in the LC. There is evidence from preclinical studies that elevated angiotensin II levels can cause neuronal damage, and that angiotensin II blockade can lead to reduction in inflammation and vascular permeability after TBI; however, studies in humans are lacking [9••, 59].
Vasopressors and Inotropes
As mentioned above, the use of vasopressors or inotropes is avoided at all costs in the LC. Based on physiologic principles of the injured brain, proponents of the LC contend that while vasopressors and inotropes raise CPP via elevations in cardiac contractility and arterial pressure, they actually can cause a decrease in perfusion to the critically injured part of the penumbra, the area of brain surrounding the primary injury. The penumbra is especially sensitive to hypoxic insults, and preserving the penumbra zone is one of the primary goals in treating secondary brain injury [9••]. Furthermore, activation of the sympathetic nervous system and catecholamine excess after TBI can lead to peripheral insults, such as Acute Respiratory Distress Syndrome (ARDS), via release of pro-inflammatory substances and increases in vascular permeability [60, 61]. Previous BTF guidelines had recommended the use of vasopressor/inotrope therapy to maintain CPP > 70 mm Hg; however, as evidence of the harmful effects of vasopressors and inotropes has accumulated, and mortality has been shown to improve with CPP < 70 mm Hg, the most recent BTF guidelines now recommend avoiding the use of vasopressors and inotropes to maintain CPP > 70 mm Hg [8].
Decompressive Surgery for Elevated ICP
Decompressive Craniectomy
Decompressive craniectomy (DC) is used to treat elevated ICP when medical management has failed. The DECRA trial (Decompressive Craniectomy in Diffuse Traumatic Brain Injury) investigated the use of early DC (within 72 h) to treat elevated ICP; of note, the study did not include those patient with mass lesions [62]. The results demonstrated that while DC was effective in treating intracranial hypertension and reducing length of ICU Stay, those who underwent DC had worse long-term neurologic outcomes as based on their Extended Glasgow Outcome Scale scores. The DECRA trial utilized a large bifrontotemporoparietal DC. The most recent BTF guidelines caution against using a bifrontal DC and, instead, recommend a large frontotemporoparietal DC [8]. This recommendation was made prior to the results of the RESCUEicp study (Randomized Evaluation of Surgery with Craniectomy for Uncontrollable Elevation of Intracranial Pressure), which evaluated the use of DC (unilateral or bilateral based on intracranial pathology and surgeon preference) in patients with refractory intracranial hypertension despite maximum medical management [63]. The results of this trial indicate that while mortality was improved in the surgical group, this was also associated with a higher incidence of vegetative state and severe disability. The rates of moderate disability or good recovery were similar between the two groups (RESCUEicp). A recent meta-analysis by Zhang et al. examined the value of DC in TBI with elevated ICP; they concluded that while DC is effective in lowering ICP and reducing mortality rates, it is associated with an increase in overall complication rates [64]. However, the benefit on long-term recovery and functional outcomes was not significant.
In the LC, DC is accepted as a life-saving maneuver for prevention of brain-stem herniation and death with uncontrolled elevations in ICP despite optimal medical treatment [18]. However, the LC advocates for treating effects of DC, specifically the increase in transcapillary hydrostatic pressure that results from the reduction in ICP. This increase in pressure leads to increased transcapillary filtration and, in turn, potentially worsening brain edema and further secondary brain injury. In order to counteract this, the LC advocates for maintaining relatively low CPP after DC with the use of antihypertensive therapy, plus albumin, as described above [9••]. A retrospective review of patients treated according to the LC demonstrated no significant difference in long-term outcome between patients who underwent DC versus those who did not have surgery, even though the patients who underwent DC had higher initial ICPs [65]. However, one must caution comparing these outcomes to DECRA and RESCUEicp, as the outcome long-term outcomes in both groups of severe TBI patients in the study by Olivecrona et al. were significantly improved compared to both the DECRA and RESCUEicp trials.
Multiple Compartment Syndrome
The equation CPP = MAP − ICP lacks a specific venous component and ignores the potential effects of venous pressure on ICP and cerebral perfusion. However, intracranial hypertension can result from increased venous pressure, both inside and outside the cranial vault [66]. Within the cranium, external compression or internal obstruction of the venous sinuses can increase venous pressure. In severe TBI patients, especially those with polytrauma, external venous compression from outside of the cranial vault can lead to increased ICP. Ill-fitted cervical collars can cause compression of the internal jugular vein, which raises ICP. Furthermore, increases in intra-abdominal pressure (IAP) or intra-thoracic pressure can raise ICP. Scalea et al. have termed this phenomenon “multiple compartment syndrome” (MCS), highlighting the relationship between the abdomen, thorax, and brain and, furthermore, the effects that treatments for one compartment may have on another [67]. Excessive fluids used to augment CPP can lead to intra-abdominal edema and increased intra-abdominal pressures, causing compression of splanchnic vasculature or abdominal compartment syndrome (ACS). Furthermore, excessive fluids can also lead to pathologic interstitial pulmonary edema, acute respiratory distress syndrome (ARDS), and increases in intra-thoracic pressure. Joseph et al. examined 17 patients with intractable intracranial hypertension, without evidence of ACS, who underwent DL as a last-resort maneuver [68]. 11 out of the 17 patients had sustained decreases in ICP and survived. Those who did not survive only had transient decreases in ICP after DL. The same group then performed a retrospective review of patients who had intractable elevation in ICP, were identified as having MCS, and underwent both DC and DL [67]. ICP was reduced consistently after both procedures. While the number of patients with MCS was small, 24 total, they had higher injury severity scores and fluid requirements compared to patients with severe TBI who underwent DC alone. Furthermore, ICP significantly decreased in all patient with MCS who underwent DL.
IAP monitoring and ACS are not included in either the BTF guidelines or LC. While not addressed in the BTF guidelines, the use of positive end-expiratory pressure (PEEP) is specified in the LC in order to prevent atelectasis and development of ARDS [9••]. Clinical data from recent years have demonstrated that PEEP does have not clinically significant effects on CPP or ICP and can be used safely in severe TBI patients [69,70,71]. This is probably related to the venous collapse within the brain, just distal to the dura, that protects against “back pressure” until central venous pressure (CVP) > ICP; flow CVP = ICP may be the basis for the efficacy of decompressive laparotomy for refractory ICP with moderate elevation in IAP. In severe TBI patients with intractable elevations in ICP—especially those with polytrauma, high fluid requirements and acute lung injury—one should consider the effects that the intra-abdominal and intra-thoracic compartments may have on intracranial pathology.
The Management of Intravascular Volume in TBI
Volume status is crucial in every critically ill patient; however, TBI patients are especially vulnerable to the effects of intravascular volume loss and replacement due to disruptions in and increased permeability of the BBB and the susceptibility of the injured brain to hypoxia. While significant research has been applied to hyperosmolar therapy for reducing ICP, there is a lack of literature and evidence about fluid management and maintenance of normovolemia after TBI.
The LC provides guidelines for the management of intravascular volume, which has been the source of much of its criticisms [72]. The main goal of fluid resuscitation in the LC is avoidance of hypovolemia via albumin supplementation and erythrocyte transfusions (which will be explored further in the succeeding sections).
The BTF guidelines, however, provide almost no guidance as to how clinicians should address fluid management in severe TBI. The most recent guidelines only state that the prior recommendation of using mannitol to reduce ICP is no longer supported, and there is insufficient evidence, thus far, to recommend hypertonic saline (HTS) over mannitol [8]. Yet, recommendations for maintenance fluid therapy, volume replacement, hemoglobin/hematocrit thresholds, and the use of blood products—all critical components in the treatment of patients with severe TBI—are lacking. Of important note, current management of trauma and damage control resuscitation has changed significantly over the last 20 years, specifically the adoption of Massive Transfusion Protocols and the 1:1:1 ratio of plasma:platelet:red blood cells (RBC) transfusion strategy in favor of crystalloid- or albumin-based initial resuscitation of trauma patients [73, 74]. Therefore, some of the findings outlined below, including the use of crystalloid-based resuscitation, are based on management strategies in trauma patients that are no longer used today.
Albumin Versus Crystalloids
The debate over the use of albumin versus crystalloid for volume replacement has been the focus of significant research and opinion in every niche of critical care; however, the literature in TBI has yet to provide any clear evidence or direction.
Post hoc analysis of subgroup of patient with TBI SAFE Study, conducted between 2001 and 2003, concluded that fluid resuscitation with albumin was associated with higher mortality rates than resuscitation with saline in critically ill TBI patients [75]. In further post hoc analysis of those TBI patients who had ICP monitors placed, the authors of the study concluded that resuscitation with albumin was associated with increased ICP and likely contributed to increased mortality during the first week post-injury [76]. These results have prompted many to shun the use of albumin in the management of TBI. However, this study has two major issues that likely preclude its applicability to current practice. First, this study was conducted during a time when the major TBI guidelines recommended maintaining CPP > 70 mm Hg, which often required the use of vasopressors as indicated in the data from the SAFE study [77]. As described above, vasopressors can lead to worsening of cerebral edema and ischemia, as well as the development of ARDS. Second, the study used a 4% albumin solution, which is hypotonic. Resuscitation with hypotonic fluids is known to worsen cerebral edema after TBI, and critics of the SAFE Study have concluded that its results only further confirmed this finding [78].
The LC guidelines continue to endorse the use of albumin in the treatment of severe TBI. Specifically, the LC recommends the use of isotonic 20% albumin in combination with crystalloids to maintain normovolemia [9••]. The use of albumin allows for decreased amount of total volume of fluid necessary to maintain normovolemia, therefore limiting further cerebral edema. However, evidence supporting the safety of albumin in TBI is limited to animal models and small, single-center studies [9••]. Furthermore, recent evidence into the effects of the endothelial glycocalyx layer in trauma signify that there is a reduced absorption effect of transcapillary oncotic pressure than would be predicted by classic Starling principles [18]. Thus, the plasma expanding capacity of albumin may be reduced in trauma and TBI patients.
Recent evidence from general critical care and trauma populations have investigated the use of saline versus balanced crystalloids, such as Lactated Ringers and Plasmalyte. Semler et al. concluded that in critically ill adults, balanced crystalloids, in comparison to normal saline, resulted in decreased mortality, need for renal replacement therapy, and persistent renal dysfunction [79]. Furthermore, higher volume of crystalloid resuscitation is associated with increased mortality [80]. However, evidence based on non-TBI populations are difficult to adopt into the management of TBI patients due to effects of BBB disruption.
Hyperosmolar Therapy
Hyperosmolar agents cause a fluid shift from the brain to the intravascular compartment, thus allowing for a reduction in ICP [81]. The mainstays of hyperosmolar therapy include mannitol and hypertonic saline (HTS), and they have become central components in the treatment of pathologic ICP after TBI [82]. However, although significant preclinical and clinical research has focused on each of these agents individually and in comparison to each other, the existing data provide inconclusive evidence to support their role in the management of TBI. The most recent BTF guidelines removed previous recommendations for the use of mannitol and, instead, stated that sufficient evidence is lacking to make any recommendation as to the use of hyperosmolar therapy [8]. Proponents of the LC contend that the hyperosmolar therapy only treats the symptom of elevated ICP, but does not have help treat the pathologic mechanism behind it. In the LC, osmotherapy is only used to treat acute prevention of brain-stem compression [9••].
Recent guidelines for fluid therapy in neurointensive care patients, produced by the European Society of Intensive Care Medicine, gave a weak recommendation for the use of hyperosmolar therapy in order to reduce ICP, specifically for ICP greater than 25 mm Hg (also a weak recommendation) [83]. However, data from 3 randomized control trials performed in TBI patients demonstrated low-quality evidence against improved outcomes with the use of hyperosmolar therapy [83]. Yet, hyperosmolar therapy will likely remain in use until significant evidence emerges that demonstrates worsened outcomes with its use. Among the two most common agents used, HTS appears to be more effective in reducing ICP burden, but this has not led to significant differences in outcomes [82, 84,85,86]. Preclinical data from Oernbo et al. also demonstrated that hyperosmolar NaCl solution induced a greater osmotic response than mannitol [87].
The results from the ongoing COBI trial (Continuous Hyperosmolar Therapy for Traumatic Brain-injured Patients; NCT03143751) may help clarify the role of HTS in acute TBI. In this multicenter, randomized control trial, patients with TBI (defined as GCS ≤ 12 and abnormal brain CT scan) will be randomized to standard care or continuous 20% HTS for at least 48 h [88]. A sodium level of greater than 155 mmol/L results in a stoppage of HTS until the next lab draw. One must be cautious in using HTS; a recent retrospective review found that hypernatremia (defined as Na > 150 mEq/L) was independently associated with increased mortality in patients with severe TBI, with higher rates of mortality in patients with severe hypernatremia (Na > 160 mEq/L) [89].
Data from preclinical experiments in animal models have demonstrated that hyperosmolar therapy reduced leukocyte recruitment at the lesion site after TBI [90]. Models of intracerebral hemorrhage and cerebral ischemia have demonstrated that HTS and mannitol may reduce pro-inflammatory mediators and pro-inflammatory microglia activation and, thereby, attenuate neuroinflammation and cerebral edema [91,92,93,94].
Blood Products
Hemoglobin and Red Blood Cell Transfusions
The current BTF guidelines have no recommendations for hemoglobin/hematocrit (Hb/Hct) thresholds or the use of blood products in resuscitation after TBI. The LC, however, promotes higher Hb/Hct thresholds and a more aggressive transfusion strategy than is the current standard in most ICUs. A recent review of the LC suggests a target Hb level of 10.5–11 g/dL, which is actually lower than previous suggested Hb target of 11.5–12 g/dL [9••]. According to proponents of the LC, the argument in favor of liberal transfusion is two-fold. Erythrocytes contribute to a significant portion of the intravascular volume; thus, at low hemoglobin concentrations, higher volumes of plasma volume expanders such as albumin and crystalloids are necessary to maintain normovolemia [9••, 18]. And given the relative hypoxia of the penumbra zone, LC proponents reason that TBI patients are particularly susceptible to anemia and hypovolemia.
However, evidence from the Transfusion Requirements in Critical Care (TRICC) Trial, specifically post hoc analysis of a subgroup of TBI patients, demonstrated no benefit from a restrictive (Hb < 7 g/dL) versus liberal (Hg < 10 g/dL) threshold for RBC transfusion [95]. Subsequent studies in TBI have confirmed this finding [96,97,98]. Maintaining Hb threshold > 10 g/dL was actually associated with higher risk of progressive hemorrhagic injury when compared to a threshold of > 7 g/dL. However, studying the effects of Hb thresholds on brain tissue oxygenation (PbtO2), increased episodes of tissue hypoxia in normal brain tissue were noted in the low Hb group, which was not seen when the monitor was placed in contused brain [96]. Yet, these results were not statistically significant, and the authors concluded that the overall data do not support the use of higher transfusion threshold in TBI.
Plasma
Traumatic injury and TBI cause disruption and damage to the endothelium, which can lead to worsening coagulopathy, vascular leak, edema, tissue injury, and inflammation [73]. Multiple studies have demonstrated the effectiveness of plasma in reducing endothelial cell permeability and reversing hypercoagulability in hemorrhagic shock [73, 99]. A recent study in severe TBI patients found significantly elevated syndecan-1 levels, a marker of endothelial glycocalyx injury, in those patients with TBI associated coagulopathy [100]. Furthermore, these patients required increased blood product transfusions and had higher risk of early mortality.
Evidence from trauma literature has demonstrated the benefit of using a balanced transfusion strategy, specifically a 1:1:1 ratio of plasma:platelet:RBC, in damage control resuscitation [74]. Post hoc analysis also revealed that TBI patients with concomitant hemorrhagic shock had worse pre-resuscitation coagulopathy than all other groups, signifying that understanding and implementing balanced damage control resuscitation is crucial for any provider caring for TBI patients [24]. Furthermore, the presence of coagulopathy has been demonstrated to lead to worsened outcomes in penetrating TBI [101].
Plasma, specifically fresh frozen plasma (FFP), is currently under investigation as a resuscitative fluid in TBI. Animal models have demonstrated improved outcomes with plasma-based resuscitation in comparison to crystalloid and colloids [102,103,•–104]. Jin et al. demonstrated that administration of FFP significantly reduced brain swelling and prevented progression of brain injury compared to treatment with NS in a swine model of TBI + hemorrhagic shock [102•]. Furthermore, Halawiesh et al. have demonstrated that lyophilized plasma provided comparable neuroprotection to FFP in a swine model of TBI, which has implications for resuscitating TBI patients in locations where access to FFP is limited, such as the combat theater [105]. Georgoff et al. demonstrated, in another swine model of TBI in combination with hemorrhagic shock and polytrauma, that resuscitation with both FFP and lyophilized plasma resulted in lower neurologic severity scores in comparison to animals resuscitated with normal saline [106].
Early plasma transfusion has been associated with improved in-hospital survival in TBI patients who presented with multifocal intracranial hemorrhage; however, this finding was not associated with other injury subtypes, including epidural hematomas, subdural hematomas, intraparenchymal contusions, or subarachnoid hemorrhage [107]. Further preclinical and clinical investigations are warranted before any definite recommendations for plasma use as a volume expander or resuscitative in TBI can be made. However, there is evidence that plasma can preserve endothelial integrity, reverse trauma-induced coagulopathy, and reduce cerebral edema and lesion size after TBI.
Conclusion
TBI is one of the least understood disorders among both the trauma and neurocritical care populations. We still have not completely elucidated the pathophysiological mechanisms that contribute to secondary brain injury. Furthermore, no therapies exist that have been proven to prevent secondary brain injury. Current practice focuses on treating ICP and CPP, in order to reduce cerebral edema, ischemia, and brain tissue damage. The management of hemodynamics and intravascular volume are crucial in TBI, but, as demonstrated in this review consensus in treatment options is lacking and many therapies require further investigation. Guidelines for the management of TBI, such as the BTF and LC, have many fundamental differences (see Table 1). However, based on the information presented, we propose that certain strategies will likely prove beneficial in the management of TBI:
For patients presenting with significant extracranial hemorrhage, resuscitation with a 1:1:1 blood product ratio should be initiated. After control of bleeding, and in those patients with isolated TBI, we propose that plasma-based volume resuscitation, as opposed to crystalloid or albumin, may improve outcomes via correction of coagulopathy, preservation of the endothelium, and reduction of intracranial hemorrhage extension. Further study is certainly warranted on the use of plasma in TBI. Once the patient has been stabilized, early and continuous beta-blocker administration is recommended to decrease the catecholamine and systemic inflammatory excess seen after TBI, which may help attenuate secondary brain injury via increase in cerebral perfusion and oxygenation, decrease in the pro-inflammatory response, and preservation of cardiac function. Alpha-2 agonist therapy has also demonstrated potential for improving outcomes after TBI. The results from the DASH after TBI Study will hopefully elucidate the efficacy of beta-blocker and alpha-2 agonist therapy in TBI (NCT01322048) [108].
Vasopressors and inotropes should be limited, and we propose that therapy should be targeted to control ICP, rather than CPP. The range of optimal CPP remains elusive, possibly because it is only a theoretical marker of CBF. What is apparent, however, is that ICP burden correlates with injury severity. When ICP is elevated, treatment should focus on reducing ICP, as opposed titrating CPP via pharmacologic augmentation of MAP. HTS is recommended to reduce acute elevations in ICP, and early DC for persistently uncontrolled ICP should be offered to patients with otherwise survivable injuries. Furthermore, the COBI trial will help provide information on early and continuous use of HTS.
Finally, although not addressed in this review, the reader should be aware of the potential benefit of cellular therapy in TBI to attenuate secondary brain injury. Preclinical and clinical data have demonstrated that stem cells—including bone marrow mononuclear cells (BMMNC), multipotent adult progenitor cells (MAPC), and mesenchymal stem cells (MSC)—decrease inflammation, cerebral edema, and improve outcomes after TBI via alteration in systemic and cerebral immune function [45, 109,110,111,112,113,114]. Currently, there are two ongoing Phase II trials evaluating autologous BMMNC in children and adults with severe TBI (NCT02525432 and NCT01851083).
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Faul M, Xu L, Wald MM, Coronado V. Traumatic brain injury in the United States. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. 2010.
Prevention CfDCa. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation. United States: Epidemiology and Rehabilitation National Center for Injury Prevention and Control; Division of Unintentional Injury Prevention Atlanta, GA. 2015.
Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13(3):171–91. https://doi.org/10.1038/nrneurol.2017.13.
Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129(4):1021–9. https://doi.org/10.1016/j.neuroscience.2004.06.046.
Kochanek PM, Dixon CE, Mondello S, Wang KKK, Lafrenaye A, Bramlett HM, et al. Multi-center pre-clinical consortia to enhance translation of therapies and biomarkers for traumatic brain injury: operation brain trauma therapy and beyond. Front Neurol. 2018;9:640. https://doi.org/10.3389/fneur.2018.00640.
Maas AIR, Menon DK, Adelson PD, Andelic N, Bell MJ, Belli A, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16(12):987–1048. https://doi.org/10.1016/s1474-4422(17)30371-x.
Caplan HW, Cox CS, Bedi SS. Do microglia play a role in sex differences in TBI? J Neurosci Res. 2017;95(1–2):509–17. https://doi.org/10.1002/jnr.23854.
Carney N, Totten AM, O’Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the management of severe traumatic brain injury. Fourth Ed Neurosurg. 2017;80(1):6–15. https://doi.org/10.1227/NEU.0000000000001432.
•• Grande PO. Critical evaluation of the lund concept for treatment of severe traumatic head injury, 25 years after its introduction. Front Neurol. 2017;8:315. https://doi.org/10.3389/fneur.2017.00315. Written by the author of the original 1992 Lund Concept manuscipt, this review outlines the physiologic basis for the components of the LC, including critical pre-clinical and clinical studies. Furthermore, the author notes changes in both the LC and BTF guidelines over the last 25 years.
Bragge P, Synnot A, Maas AI, Menon DK, Cooper DJ, Rosenfeld JV, et al. A state-of-the-science overview of randomized controlled trials evaluating acute management of moderate-to-severe traumatic brain injury. J Neurotrauma. 2016;33(16):1461–78. https://doi.org/10.1089/neu.2015.4233.
Nordstrom CH, Koskinen LO, Olivecrona M. Aspects on the physiological and biochemical foundations of neurocritical care. Front Neurol. 2017;8:274. https://doi.org/10.3389/fneur.2017.00274.
Rangel-Castilla L, Gasco J, Nauta HJ, Okonkwo DO, Robertson CS. Cerebral pressure autoregulation in traumatic brain injury. Neurosurg Focus. 2008;25(4):E7. https://doi.org/10.3171/FOC.2008.25.10.E7.
Chesnut RM, Temkin N, Carney N, Dikmen S, Rondina C, Videtta W, et al. A trial of intracranial-pressure monitoring in traumatic brain injury. N Engl J Med. 2012;367(26):2471–81. https://doi.org/10.1056/NEJMoa1207363.
Talving P, Karamanos E, Teixeira PG, Skiada D, Lam L, Belzberg H, et al. Intracranial pressure monitoring in severe head injury: compliance with Brain Trauma Foundation guidelines and effect on outcomes: a prospective study. J Neurosurg. 2013;119(5):1248–54. https://doi.org/10.3171/2013.7.JNS122255.
Gerber LM, Chiu YL, Carney N, Hartl R, Ghajar J. Marked reduction in mortality in patients with severe traumatic brain injury. J Neurosurg. 2013;119(6):1583–90. https://doi.org/10.3171/2013.8.JNS13276.
Farahvar A, Gerber LM, Chiu YL, Carney N, Hartl R, Ghajar J. Increased mortality in patients with severe traumatic brain injury treated without intracranial pressure monitoring. J Neurosurg. 2012;117(4):729–34. https://doi.org/10.3171/2012.7.JNS111816.
• Guiza F, Depreitere B, Piper I, Citerio G, Chambers I, Jones PA et al. Visualizing the pressure and time burden of intracranial hypertension in adult and paediatric traumatic brain injury. Intensive Care Med. 2015;41(6):1067–76. https://doi.org/10.1007/s00134-015-3806-1. This clinical trial effectively demonstrates the importance of the concept of ICP burden: that time spent over a critical ICP threshold leads to poor outcomes after TBI.
Koskinen LO, Olivecrona M, Grande PO. Severe traumatic brain injury management and clinical outcome using the Lund concept. Neuroscience. 2014;283:245–55. https://doi.org/10.1016/j.neuroscience.2014.06.039.
Guiza F, Meyfroidt G, Piper I, Citerio G, Chambers I, Enblad P, et al. Cerebral perfusion pressure insults and associations with outcome in adult traumatic brain injury. J Neurotrauma. 2017;34(16):2425–31. https://doi.org/10.1089/neu.2016.4807.
Hutchinson PJ, Jalloh I, Helmy A, Carpenter KL, Rostami E, Bellander BM, et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med. 2015;41(9):1517–28. https://doi.org/10.1007/s00134-015-3930-y.
Andresen M, Donnelly J, Aries M, Juhler M, Menon D, Hutchinson P, et al. Further controversies about brain tissue oxygenation pressure-reactivity after traumatic brain injury. Neurocrit Care. 2018;28(2):162–8. https://doi.org/10.1007/s12028-017-0438-z.
Pigula FA, Wald SL, Shackford SR, Vane DW. The effect of hypotension and hypoxia on children with severe head injuries. J Pediatr Surg. 1993;28(3):310–4 discussion 5-6.
Spaite DW, Hu C, Bobrow BJ, Chikani V, Barnhart B, Gaither JB, et al. Association of out-of-hospital hypotension depth and duration with traumatic brain injury mortality. Ann Emerg Med. 2017;70(4):522-30 e1. https://doi.org/10.1016/j.annemergmed.2017.03.027.
Galvagno SM Jr, Fox EE, Appana SN, Baraniuk S, Bosarge PL, Bulger EM, et al. Outcomes after concomitant traumatic brain injury and hemorrhagic shock: a secondary analysis from the Pragmatic, Randomized Optimal Platelets and Plasma Ratios trial. J Trauma Acute Care Surg. 2017;83(4):668–74. https://doi.org/10.1097/TA.0000000000001584.
Bogert JN, Harvin JA, Cotton BA. Damage control resuscitation. J Intensive Care Med. 2016;31(3):177–86. https://doi.org/10.1177/0885066614558018.
Krishnamoorthy V, Chaikittisilpa N, Kiatchai T, Vavilala M. Hypertension after severe traumatic brain injury: friend or foe? J Neurosurg Anesthesiol. 2017;29(4):382–7. https://doi.org/10.1097/ANA.0000000000000370.
Hawkins WE, Clower BR. Myocardial damage after head trauma and simulated intracranial haemorrhage in mice: the role of the autonomic nervous system. Cardiovasc Res. 1971;5(4):524–9.
Clifton GL, Ziegler MG, Grossman RG. Circulating catecholamines and sympathetic activity after head injury. Neurosurgery. 1981;8(1):10–4.
Woolf PD, Hamill RW, Lee LA, Cox C, McDonald JV. The predictive value of catecholamines in assessing outcome in traumatic brain injury. J Neurosurg. 1987;66(6):875–82. https://doi.org/10.3171/jns.1987.66.6.0875.
Rizoli SB, Jaja BN, Di Battista AP, Rhind SG, Neto AC, da Costa L, et al. Catecholamines as outcome markers in isolated traumatic brain injury: the COMA-TBI study. Crit Care. 2017;21(1):37. https://doi.org/10.1186/s13054-017-1620-6.
Krishnamoorthy V, Mackensen GB, Gibbons EF, Vavilala MS. Cardiac dysfunction after neurologic injury: what do we know and where are we going? Chest. 2016;149(5):1325–31. https://doi.org/10.1016/j.chest.2015.12.014.
Meyfroidt G, Baguley IJ, Menon DK. Paroxysmal sympathetic hyperactivity: the storm after acute brain injury. Lancet Neurol. 2017;16(9):721–9. https://doi.org/10.1016/s1474-4422(17)30259-4.
Di Battista AP, Rhind SG, Hutchison MG, Hassan S, Shiu MY, Inaba K, et al. Inflammatory cytokine and chemokine profiles are associated with patient outcome and the hyperadrenergic state following acute brain injury. J Neuroinflamm. 2016;13:40. https://doi.org/10.1186/s12974-016-0500-3.
Chaikittisilpa N, Krishnamoorthy V, Lele AV, Qiu Q, Vavilala MS. Characterizing the relationship between systemic inflammatory response syndrome and early cardiac dysfunction in traumatic brain injury. J Neurosci Res. 2018;96(4):661–70. https://doi.org/10.1002/jnr.24100.
Krishnamoorthy V, Rowhani-Rahbar A, Chaikittisilpa N, Gibbons EF, Rivara FP, Temkin NR, et al. Association of early hemodynamic profile and the development of systolic dysfunction following traumatic brain injury. Neurocrit Care. 2017;26(3):379–87. https://doi.org/10.1007/s12028-016-0335-x.
Asgeirsson B, Grande PO, Nordstrom CH. A new therapy of post-trauma brain oedema based on haemodynamic principles for brain volume regulation. Intensive Care Med. 1994;20(4):260–7.
Grande PO. The “Lund Concept” for the treatment of severe head trauma–physiological principles and clinical application. Intensive Care Med. 2006;32(10):1475–84. https://doi.org/10.1007/s00134-006-0294-3.
Zoerle T, Carbonara M, Zanier ER, Ortolano F, Bertani G, Magnoni S, et al. Rethinking neuroprotection in severe traumatic brain injury: toward bedside neuroprotection. Front Neurol. 2017;8:354. https://doi.org/10.3389/fneur.2017.00354.
Stocchetti N, Maas AI. Traumatic intracranial hypertension. N Engl J Med. 2014;370(22):2121–30. https://doi.org/10.1056/NEJMra1208708.
Alali AS, Mukherjee K, McCredie VA, Golan E, Shah PS, Bardes JM, et al. Beta-blockers and traumatic brain injury: a systematic review, meta-analysis, and Eastern Association for the Surgery of Trauma Guideline. Ann Surg. 2017;266(6):952–61. https://doi.org/10.1097/SLA.0000000000002286.
Ley EJ, Park R, Dagliyan G, Palestrant D, Miller CM, Conti PS, et al. In vivo effect of propranolol dose and timing on cerebral perfusion after traumatic brain injury. J Trauma. 2010;68(2):353–6. https://doi.org/10.1097/TA.0b013e3181c8269a.
Ley EJ, Clond MA, Bukur M, Park R, Chervonski M, Dagliyan G, et al. beta-adrenergic receptor inhibition affects cerebral glucose metabolism, motor performance, and inflammatory response after traumatic brain injury. J Trauma Acute Care Surg. 2012;73(1):33–40. https://doi.org/10.1097/TA.0b013e31825a769b.
Liou DZ, Ko A, Volod O, Barmparas G, Harada MY, Martin MJ, et al. Thromboelastography after murine TBI and implications of beta-adrenergic receptor knockout. Neurocrit Care. 2016;25(1):145–52. https://doi.org/10.1007/s12028-015-0223-9.
Wang J, Li J, Sheng X, Zhao H, Cao XD, Wang YQ, et al. Beta-adrenoceptor mediated surgery-induced production of pro-inflammatory cytokines in rat microglia cells. J Neuroimmunol. 2010;223(1–2):77–83. https://doi.org/10.1016/j.jneuroim.2010.04.006.
Kota DJ, Prabhakara KS, van Brummen AJ, Bedi S, Xue H, DiCarlo B, et al. Propranolol and mesenchymal stromal cells combine to treat traumatic brain injury. Stem Cells Transl Med. 2016;5(1):33–44. https://doi.org/10.5966/sctm.2015-0065.
Genet GF, Bentzer P, Ostrowski SR, Johansson PI. Resuscitation with pooled and pathogen-reduced plasma attenuates the increase in brain water content following traumatic brain injury and hemorrhagic shock in rats. J Neurotrauma. 2017;34(5):1054–62. https://doi.org/10.1089/neu.2016.4574.
Arbabi S, Campion EM, Hemmila MR, Barker M, Dimo M, Ahrns KS, et al. Beta-blocker use is associated with improved outcomes in adult trauma patients. J Trauma. 2007;62(1):56–61. https://doi.org/10.1097/ta.0b013e31802d972b discussion-2.
Cotton BA, Snodgrass KB, Fleming SB, Carpenter RO, Kemp CD, Arbogast PG, et al. Beta-blocker exposure is associated with improved survival after severe traumatic brain injury. J Trauma. 2007;62(1):26–33. https://doi.org/10.1097/ta.0b013e31802d02d0 discussion-5.
Inaba K, Teixeira PG, David JS, Chan LS, Salim A, Brown C, et al. Beta-blockers in isolated blunt head injury. J Am Coll Surg. 2008;206(3):432–8. https://doi.org/10.1016/j.jamcollsurg.2007.10.005.
Zangbar B, Khalil M, Rhee P, Joseph B, Kulvatunyou N, Tang A, et al. Metoprolol improves survival in severe traumatic brain injury independent of heart rate control. J Surg Res. 2016;200(2):586–92. https://doi.org/10.1016/j.jss.2015.08.020.
Ko A, Harada MY, Barmparas G, Thomsen GM, Alban RF, Bloom MB, et al. Early propranolol after traumatic brain injury is associated with lower mortality. J Trauma Acute Care Surg. 2016;80(4):637–42. https://doi.org/10.1097/TA.0000000000000959.
Ahl R, Thelin EP, Sjolin G, Bellander BM, Riddez L, Talving P, et al. beta-Blocker after severe traumatic brain injury is associated with better long-term functional outcome: a matched case control study. Eur J Trauma Emerg Surg. 2017;43(6):783–9. https://doi.org/10.1007/s00068-017-0779-5.
Murry JS, Hoang DM, Barmparas G, Harada MY, Bukur M, Bloom MB, et al. Prospective evaluation of early propranolol after traumatic brain injury. J Surg Res. 2016;200(1):221–6. https://doi.org/10.1016/j.jss.2015.06.045.
Bible LE, Pasupuleti LV, Alzate WD, Gore AV, Song KJ, Sifri ZC, et al. Early propranolol administration to severely injured patients can improve bone marrow dysfunction. J Trauma Acute Care Surg. 2014;77(1):54–60. https://doi.org/10.1097/ta.0000000000000264 discussion 59-60.
Aryan HE, Box KW, Ibrahim D, Desiraju U, Ames CP. Safety and efficacy of dexmedetomidine in neurosurgical patients. Brain Inj. 2006;20(8):791–8. https://doi.org/10.1080/02699050600789447.
Schoeler M, Loetscher PD, Rossaint R, Fahlenkamp AV, Eberhardt G, Rex S, et al. Dexmedetomidine is neuroprotective in an in vitro model for traumatic brain injury. BMC Neurol. 2012;12:20. https://doi.org/10.1186/1471-2377-12-20.
Hoffman WE, Cheng MA, Thomas C, Baughman VL, Albrecht RF. Clonidine decreases plasma catecholamines and improves outcome from incomplete ischemia in the rat. Anesth Analg. 1991;73(4):460–4.
Humble SS, Wilson LD, Leath TC, Marshall MD, Sun DZ, Pandharipande PP, et al. ICU sedation with dexmedetomidine after severe traumatic brain injury. Brain Inj. 2016;30(10):1266–70. https://doi.org/10.1080/02699052.2016.1187289.
Ruiz-Ortega M, Lorenzo O, Suzuki Y, Ruperez M, Egido J. Proinflammatory actions of angiotensins. Curr Opin Nephrol Hypertens. 2001;10(3):321–9.
Contant CF, Valadka AB, Gopinath SP, Hannay HJ, Robertson CS. Adult respiratory distress syndrome: a complication of induced hypertension after severe head injury. J Neurosurg. 2001;95(4):560–8. https://doi.org/10.3171/jns.2001.95.4.0560.
Davison DL, Terek M, Chawla LS. Neurogenic pulmonary edema. Crit Care. 2012;16(2):212. https://doi.org/10.1186/cc11226.
Cooper DJ, Rosenfeld JV, Murray L, Arabi YM, Davies AR, D’Urso P, et al. Decompressive craniectomy in diffuse traumatic brain injury. N Engl J Med. 2011;364(16):1493–502. https://doi.org/10.1056/NEJMoa1102077.
Hutchinson PJ, Kolias AG, Timofeev IS, Corteen EA, Czosnyka M, Timothy J, et al. Trial of decompressive craniectomy for traumatic intracranial hypertension. N Engl J Med. 2016;375(12):1119–30. https://doi.org/10.1056/NEJMoa1605215.
Zhang D, Xue Q, Chen J, Dong Y, Hou L, Jiang Y, et al. Decompressive craniectomy in the management of intracranial hypertension after traumatic brain injury: a systematic review and meta-analysis. Sci Rep. 2017;7(1):8800. https://doi.org/10.1038/s41598-017-08959-y.
Olivecrona M, Rodling-Wahlstrom M, Naredi S, Koskinen LO. Effective ICP reduction by decompressive craniectomy in patients with severe traumatic brain injury treated by an ICP-targeted therapy. J Neurotrauma. 2007;24(6):927–35. https://doi.org/10.1089/neu.2005.356E.
Wilson MH. Monro-Kellie 2.0: the dynamic vascular and venous pathophysiological components of intracranial pressure. J Cereb Blood Flow Metab. 2016;36(8):1338–50. https://doi.org/10.1177/0271678x16648711.
Scalea TM, Bochicchio GV, Habashi N, McCunn M, Shih D, McQuillan K, et al. Increased intra-abdominal, intrathoracic, and intracranial pressure after severe brain injury: multiple compartment syndrome. J Trauma. 2007;62(3):647–56. https://doi.org/10.1097/ta.0b013e31802ee542 discussion 56.
Joseph DAK, Dutton RP, Aarabi B, Scalea TM. Decompressive laparotomy to treat intractable intracranial hypertension after traumatic brain injury. J Trauma: Injury Infect Crit Care. 2004;57(4):687–95. https://doi.org/10.1097/01.Ta.0000140645.84897.F2.
Asehnoune K, Roquilly A, Cinotti R. Respiratory management in patients with severe brain injury. Crit Care. 2018;22(1):76. https://doi.org/10.1186/s13054-018-1994-0.
Boone MD, Jinadasa SP, Mueller A, Shaefi S, Kasper EM, Hanafy KA, et al. The effect of positive end-expiratory pressure on intracranial pressure and cerebral hemodynamics. Neurocrit Care. 2017;26(2):174–81. https://doi.org/10.1007/s12028-016-0328-9.
Nemer SN, Caldeira JB, Santos RG, Guimaraes BL, Garcia JM, Prado D, et al. Effects of positive end-expiratory pressure on brain tissue oxygen pressure of severe traumatic brain injury patients with acute respiratory distress syndrome: a pilot study. J Crit Care. 2015;30(6):1263–6. https://doi.org/10.1016/j.jcrc.2015.07.019.
Sharma D, Vavilala MS. Lund concept for the management of traumatic brain injury: a physiological principle awaiting stronger evidence. J Neurosurg Anesthesiol. 2011;23(4):363–7. https://doi.org/10.1097/01.ana.0000405613.27980.ea.
Holcomb JB, Pati S. Optimal trauma resuscitation with plasma as the primary resuscitative fluid: the surgeon’s perspective. Hematol Am Soc Hematol Educ Progr. 2013;2013:656–9. https://doi.org/10.1182/asheducation-2013.1.656.
Holcomb JB, Tilley BC, Baraniuk S, Fox EE, Wade CE, Podbielski JM, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA. 2015;313(5):471–82. https://doi.org/10.1001/jama.2015.12.
Finfer S, Norton R, Bellomo R, Boyce N, French J, Myburgh J. The SAFE study: saline vs albumin for fluid resuscitation in the critically ill. Vox Sang. 2004;87(Suppl 2):123–31. https://doi.org/10.1111/j.1741-6892.2004.00468.x.
Cooper DJ, Myburgh J, Heritier S, Finfer S, Bellomo R, Billot L, et al. Albumin resuscitation for traumatic brain injury: is intracranial hypertension the cause of increased mortality? J Neurotrauma. 2013;30(7):512–8. https://doi.org/10.1089/neu.2012.2573.
The Brain Trauma Foundation. The American Association of Neurological Surgeons. The Joint Section on Neurotrauma and Critical Care. Guidelines for cerebral perfusion pressure. J Neurotrauma. 2000;17(6-7):507–11. https://doi.org/10.1089/neu.2000.17.507.
Ertmer C, Van Aken H. Fluid therapy in patients with brain injury: what does physiology tell us? Crit Care. 2014;18(2):119. https://doi.org/10.1186/cc13764.
Semler MW, Self WH, Wanderer JP, Ehrenfeld JM, Wang L, Byrne DW, et al. Balanced crystalloids versus saline in critically Ill adults. N Engl J Med. 2018;378(9):829–39. https://doi.org/10.1056/NEJMoa1711584.
Ko A, Harada MY, Barmparas G, Smith EJT, Birch K, Barnard ZR, et al. Limit crystalloid resuscitation after traumatic brain injury. Am Surg. 2017;83(12):1447–52.
Tyagi R, Donaldson K, Loftus CM, Jallo J. Hypertonic saline: a clinical review. Neurosurg Rev. 2007;30(4):277–89. https://doi.org/10.1007/s10143-007-0091-7 discussion 89-90.
Mangat HS. Hypertonic saline infusion for treating intracranial hypertension after severe traumatic brain injury. Crit Care. 2018;22(1):37. https://doi.org/10.1186/s13054-018-1963-7.
Oddo M, Poole D, Helbok R, Meyfroidt G, Stocchetti N, Bouzat P, et al. Fluid therapy in neurointensive care patients: ESICM consensus and clinical practice recommendations. Intensive Care Med. 2018;44(4):449–63. https://doi.org/10.1007/s00134-018-5086-z.
Berger-Pelleiter E, Emond M, Lauzier F, Shields JF, Turgeon AF. Hypertonic saline in severe traumatic brain injury: a systematic review and meta-analysis of randomized controlled trials. CJEM. 2016;18(2):112–20. https://doi.org/10.1017/cem.2016.12.
Alnemari AM, Krafcik BM, Mansour TR, Gaudin D. A comparison of pharmacologic therapeutic agents used for the reduction of intracranial pressure after traumatic brain injury. World Neurosurg. 2017;106:509–28. https://doi.org/10.1016/j.wneu.2017.07.009.
Burgess S, Abu-Laban RB, Slavik RS, Vu EN, Zed PJ. A systematic review of randomized controlled trials comparing hypertonic sodium solutions and mannitol for traumatic brain injury: implications for emergency department management. Ann Pharmacother. 2016;50(4):291–300. https://doi.org/10.1177/1060028016628893.
Oernbo EK, Lykke K, Steffensen AB, Tollner K, Kruuse C, Rath MF, et al. Cerebral influx of Na(+) and Cl(−) as the osmotherapy-mediated rebound response in rats. Fluids Barriers CNS. 2018;15(1):27. https://doi.org/10.1186/s12987-018-0111-8.
Roquilly A, Lasocki S, Moyer JD, Huet O, Perrigault PF, Dahyot-Fizelier C, et al. COBI (COntinuous hyperosmolar therapy for traumatic Brain-Injured patients) trial protocol: a multicentre randomised open-label trial with blinded adjudication of primary outcome. BMJ Open. 2017;7(9):e018035. https://doi.org/10.1136/bmjopen-2017-018035.
Vedantam A, Robertson CS, Gopinath SP. Morbidity and mortality associated with hypernatremia in patients with severe traumatic brain injury. Neurosurg Focus. 2017;43(5):E2. https://doi.org/10.3171/2017.7.FOCUS17418.
Kumasaka K, Marks JA, Eisenstadt R, Murcy MA, Samadi D, Li S, et al. In vivo leukocyte-mediated brain microcirculatory inflammation: a comparison of osmotherapies and progesterone in severe traumatic brain injury. Am J Surg. 2014;208(6):961–8. https://doi.org/10.1016/j.amjsurg.2014.08.004 discussion 7-8.
Schreibman DL, Hong CM, Keledjian K, Ivanova S, Tsymbalyuk S, Gerzanich V, et al. Mannitol and hypertonic saline reduce swelling and modulate inflammatory markers in a rat model of intracerebral hemorrhage. Neurocrit Care. 2018;29(2):253–63. https://doi.org/10.1007/s12028-018-0535-7.
Huang LQ, Zhu GF, Deng YY, Jiang WQ, Fang M, Chen CB, et al. Hypertonic saline alleviates cerebral edema by inhibiting microglia-derived TNF-alpha and IL-1beta-induced Na-K-Cl Cotransporter up-regulation. J Neuroinflamm. 2014;11:102. https://doi.org/10.1186/1742-2094-11-102.
Zeng WX, Han YL, Zhu GF, Huang LQ, Deng YY, Wang QS, et al. Hypertonic saline attenuates expression of Notch signaling and proinflammatory mediators in activated microglia in experimentally induced cerebral ischemia and hypoxic BV-2 microglia. BMC Neurosci. 2017;18(1):32. https://doi.org/10.1186/s12868-017-0351-6.
Wen M, Ye J, Han Y, Huang L, Yang H, Jiang W, et al. Hypertonic saline regulates microglial M2 polarization via miR-200b/KLF4 in cerebral edema treatment. Biochem Biophys Res Commun. 2018;499(2):345–53. https://doi.org/10.1016/j.bbrc.2018.03.161.
McIntyre LA, Fergusson DA, Hutchison JS, Pagliarello G, Marshall JC, Yetisir E, et al. Effect of a liberal versus restrictive transfusion strategy on mortality in patients with moderate to severe head injury. Neurocrit Care. 2006;5(1):4–9.
Yamal JM, Rubin ML, Benoit JS, Tilley BC, Gopinath S, Hannay HJ, et al. Effect of hemoglobin transfusion threshold on cerebral hemodynamics and oxygenation. J Neurotrauma. 2015;32(16):1239–45. https://doi.org/10.1089/neu.2014.3752.
Carr KR, Rodriguez M, Ottesen A, Michalek J, Son C, Patel V, et al. Association between relative anemia and early functional recovery after severe traumatic brain injury (TBI). Neurocrit Care. 2016;25(2):185–92. https://doi.org/10.1007/s12028-016-0273-7.
Robertson CS, Hannay HJ, Yamal JM, Gopinath S, Goodman JC, Tilley BC, et al. Effect of erythropoietin and transfusion threshold on neurological recovery after traumatic brain injury: a randomized clinical trial. JAMA. 2014;312(1):36–47. https://doi.org/10.1001/jama.2014.6490.
Cardenas JC, Cap AP, Swartz MD, Huby Mdel P, Baer LA, Matijevic N, et al. Plasma resuscitation promotes coagulation homeostasis following shock-induced hypercoagulability. Shock. 2016;45(2):166–73. https://doi.org/10.1097/SHK.0000000000000504.
Albert V, Subramanian A, Agrawal D, Pati HP, Gupta SD, Mukhopadhyay AK. Acute traumatic endotheliopathy in isolated severe brain injury and its impact on clinical outcome. Med Sci. 2018;6(1):2. https://doi.org/10.3390/medsci6010005.
Folkerson LE, Sloan D, Davis E, Kitagawa RS, Cotton BA, Holcomb JB, et al. Coagulopathy as a predictor of mortality after penetrating traumatic brain injury. Am J Emerg Med. 2018;36(1):38–42. https://doi.org/10.1016/j.ajem.2017.06.057.
• Jin G, DeMoya MA, Duggan M, Knightly T, Mejaddam AY, Hwabejire J et al. Traumatic brain injury and hemorrhagic shock: evaluation of different resuscitation strategies in a large animal model of combined insults. Shock. 2012;38(1):49–56. https://doi.org/10.1097/shk.0b013e3182574778. This pre-clinical study, as well as other from the same group below, demonstrates the potential for plasma-based resuscitation to reduce hemorrhage progression and improve outcomes after TBI.
Halaweish I, Bambakidis T, He W, Linzel D, Chang Z, Srinivasan A, et al. Early resuscitation with fresh frozen plasma for traumatic brain injury combined with hemorrhagic shock improves neurologic recovery. J Am Coll Surg. 2015;220(5):809–19. https://doi.org/10.1016/j.jamcollsurg.2015.01.057.
Hwabejire JO, Imam AM, Jin G, Liu B, Li Y, Sillesen M, et al. Differential effects of fresh frozen plasma and normal saline on secondary brain damage in a large animal model of polytrauma, hemorrhage and traumatic brain injury. J Trauma Acute Care Surg. 2013;75(6):968–74. https://doi.org/10.1097/ta.0b013e31829a021a discussion 74-5.
Halaweish I, Bambakidis T, Nikolian VC, Georgoff P, Bruhn P, Piascik P, et al. Early resuscitation with lyophilized plasma provides equal neuroprotection compared with fresh frozen plasma in a large animal survival model of traumatic brain injury and hemorrhagic shock. J Trauma Acute Care Surg. 2016;81(6):1080–7. https://doi.org/10.1097/TA.0000000000001204.
Georgoff PE, Nikolian VC, Halaweish I, Chtraklin K, Bruhn PJ, Eidy H, et al. Resuscitation with lyophilized plasma is safe and improves neurological recovery in a long-term survival model of swine subjected to traumatic brain injury, hemorrhagic shock, and polytrauma. J Neurotrauma. 2017;34(13):2167–75. https://doi.org/10.1089/neu.2016.4859.
Chang R, Folkerson LE, Sloan D, Tomasek JS, Kitagawa RS, Choi HA, et al. Early plasma transfusion is associated with improved survival after isolated traumatic brain injury in patients with multifocal intracranial hemorrhage. Surgery. 2017;161(2):538–45. https://doi.org/10.1016/j.surg.2016.08.023.
Patel MB, McKenna JW, Alvarez JM, Sugiura A, Jenkins JM, Guillamondegui OD, et al. Decreasing adrenergic or sympathetic hyperactivity after severe traumatic brain injury using propranolol and clonidine (DASH After TBI Study): study protocol for a randomized controlled trial. Trials. 2012;13:177. https://doi.org/10.1186/1745-6215-13-177.
Bedi SS, Hetz R, Thomas C, Smith P, Olsen AB, Williams S, et al. Intravenous multipotent adult progenitor cell therapy attenuates activated microglial/macrophage response and improves spatial learning after traumatic brain injury. Stem Cells Transl Med. 2013;2(12):953–60. https://doi.org/10.5966/sctm.2013-0100.
Bedi SS, Aertker BM, Liao GP, Caplan HW, Bhattarai D, Mandy F, et al. Therapeutic time window of multipotent adult progenitor therapy after traumatic brain injury. J Neuroinflamm. 2018;15(1):84. https://doi.org/10.1186/s12974-018-1122-8.
Cox CS Jr, Baumgartner JE, Harting MT, Worth LL, Walker PA, Shah SK, et al. Autologous bone marrow mononuclear cell therapy for severe traumatic brain injury in children. Neurosurgery. 2011;68(3):588–600. https://doi.org/10.1227/NEU.0b013e318207734c.
Jackson ML, Srivastava AK, Cox CS Jr. Preclinical progenitor cell therapy in traumatic brain injury: a meta-analysis. J Surg Res. 2017;214:38–48. https://doi.org/10.1016/j.jss.2017.02.078.
Liao GP, Harting MT, Hetz RA, Walker PA, Shah SK, Corkins CJ, et al. Autologous bone marrow mononuclear cells reduce therapeutic intensity for severe traumatic brain injury in children. Pediatr Crit Care Med. 2015;16(3):245–55. https://doi.org/10.1097/PCC.0000000000000324.
Cox CS Jr, Hetz RA, Liao GP, Aertker BM, Ewing-Cobbs L, Juranek J, et al. Treatment of severe adult traumatic brain injury using bone marrow mononuclear cells. Stem Cells. 2017;35(4):1065–79. https://doi.org/10.1002/stem.2538.
Author information
Authors and Affiliations
Contributions
Conception, design, drafting of manuscript: HWC and CSC. Critical revision of the manuscript for important intellectual content: HWC and CSC. Manuscript supervision: HWC and CSC.
Corresponding author
Ethics declarations
Conflict of interest
Henry W. Caplan declares no potential conflicts of interest. Charles S. Cox Jr holds equity and royalty interests in Cellvation Inc. as well as a sponsored research agreement and scientific advisory board position. Dr. Cox Jr has sponsored research and holds a scientific advisory board position for CBR, Inc. He also sponsored research for Hope Bio, Inc., as well as Sponsored research and received equity/royalty interest from Coagulex, Inc.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical collection on Traumatic Brain Injury Surgery.
Rights and permissions
About this article

Cite this article
Caplan, H.W., Cox, C.S. Resuscitation Strategies for Traumatic Brain Injury. Curr Surg Rep 7, 14 (2019). https://doi.org/10.1007/s40137-019-0237-x
Published:
DOI: https://doi.org/10.1007/s40137-019-0237-x