
Abstract
Purpose of Review
This review aims to briefly discuss the new paradigms explored in the recent past especially for the diagnosis and treatment of hypersensitivity pneumonitis (HP).
Recent Findings
HP has now been classified into non-fibrotic and fibrotic phenotypes. The diagnosis of non-fibrotic and fibrotic HP has been categorized into “typical,” “compatible,” and “indeterminate” in terms of radiology and histopathology, grading the degree of diagnostic confidence. The newly explored role of antifibrotic drugs provides a treatment prospect for patients with fibrotic HP whose options were largely limited to corticosteroids and immunosuppressants.
Summary
The official guidelines provide us with a systematic approach towards making a confident diagnosis of HP and its subtypes. Recent pharmacological studies have enlightened our knowledge with a variety of treatment options, especially for fibrotic HP. However, a multitude of questions still remain unanswered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypersensitivity pneumonitis (HP) is an immunologically mediated disease affecting the lung parenchyma and small airways in genetically susceptible individuals, when exposed to a known or an unknown inhaled antigen.
Campbell diagnosed the first case of HP in farmers, more than 100 years ago [1]. Its etio-pathogenesis was first described in the early 1960s by Dr. Jack Pepys et al. [2].
HP was traditionally classified into acute, subacute, and chronic subtypes, depending on the duration of symptoms and the intensity and frequency of exposure to trigger agents [3]. Acute HP has been attributed to occur hours to days following a heavy exposure to an inciting agent, while chronic HP occurs secondary to persistent, low level of exposure to a trigger over a prolonged time period. Literature does not clearly demarcate the subacute HP phenotype, owing to an undefined time interval and overlapping radiological features. It occurs due to a more prolonged exposure to the offending agent than that seen in acute HP, generally in weeks to months with clinical presentation ranging in between the above two entities [4]. Subacute HP has been postulated to fall in the spectrum of acute HP [5]. Due to difficulty in identifying the subacute HP phenotype with no major practical implications, it was suggested to categorize HP into acute/cellular and chronic/fibrotic subtypes [6]. The latest guidelines now classify HP into non fibrotic (nfHP) and fibrotic HP (fHP) subtypes with an arbitrary cut-off duration of 6 months [7••, 8••]. ‘Cryptogenic HP’ is an entity described in cases with clinicoradiological features typical of HP, where the offending antigen remains unrecognized despite a thorough search for the same. The failure to recognize the offending trigger leads to a subsequent ongoing exposure to the occult antigen which thus makes the disease take a chronic course, similar to idiopathic pulmonary fibrosis (IPF) [6, 8••].
We have come a long way in understanding the disease mechanism, identifying inciting antigens, its presentation, diagnostic modalities, and treatment strategies. This review aims to briefly discuss the new paradigms explored in the recent past especially for the diagnosis and treatment of HP.
Epidemiology
Prevalence of HP has significantly varied over time, as reported by different ILD registries across the globe. Dated back to 2001, data from the registries of 3 European countries (Belgium, Germany, and Italy) suggested HP contributed to 4–15% of all ILD cases [9]. Recent registries have also reported wide variations in their results. The 2015 EXCITING-ILD registry reported 18% of their cases as HP [10] while HP contributed to 9.4% of all ILDs in an Australian cohort [11]. The multicenter Indian ILD registry observed that HP accounted for 47.3% of all ILD cases, with a considerable variation across its centers in the prevalence of HP [12]. Data from another single-center prospective study from India suggested that HP contributed to 10.7% of all ILDs [13]. There has been a considerable heterogeneity in the prevalence of HP. A recent American cohort analysis suggested the 1-year prevalence rates for HP ranged from 1.67 to 2.71 per 100,000 persons and 1-year cumulative incidence rates from 1.28 to 1.94 per 100,000 persons [14]. A Danish registry reported an average HP incidence of 1.16 per 100,000 citizens [15]. In a recent study from India, the crude annual incidence rate and the prevalence (per 100,000 population) of HP was estimated to be 1.4–2.9 and 6.2–12.3, respectively [16]. The possible explanation to the varied incidence and prevalence is the diverse and complex environmental, socio-cultural, economic, and genetic milieu which the subjects are exposed to across the world. A lack of uniform diagnostic criteria across the various registries with different study designs further contribute to the heterogeneity.
Pathogenesis
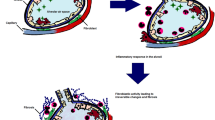
It has been postulated that patients with certain genetic mutations, defined as the “first hit” when exposed to the inciting trigger factor which acts as the “second hit,” develop the disease [4]. Certain agents like viral infections, pesticide exposure, and pollutants may act as “facilitators” for the second hit (Fig. 1) [17–19]. Air pollution was found to act as a facilitator for development of HP, a 7% higher odds of developing HP was noted for every 10-mcg m−3 increase in level of particulate matter < 2.5 mcg in diameter [19].

Pathogenesis of hypersensitivity pneumonitis
An exhaustive list of potentially offending antigens has evolved over the years. Microorganisms (like bacteria, protozoa, fungi), animal proteins, chemicals (like isocyanates, anhydrides), and feather duvets all have been implicated in causing HP [20–23]. The effect of cigarette smoke on HP depends on the duration of exposure. Short-duration smoking has been proposed to be protective, while smoking for longer duration accelerates lung inflammation and fibrosis in animal models [24]. Certain unique and inimitable exposures have also been implicated in predisposing to HP which includes dug wells and musical instruments [25, 26].
Diagnosis
An array of diagnostic modalities have been used in previous studies ranging from radiology, BAL fluid analyses, and lung biopsies. Some exhaustive diagnostic models have also been proposed over the years; however, they are limited by methodology and validation [27–30]. Although HP as a disease entity has been known since decades, the first official diagnostic guidelines came into force in 2020 by the ATS/JRS/ALAT society followed by the CHEST 2021 guidelines [7••, 8••].
The application of combinations of multiple modalities for the diagnosis of HP was endorsed by both. Features like history of exposure to an offending antigen, bronchoalveolar lavage (BAL) lymphocytosis in the background of a typical radiology and/or biopsy were considered to be consistent with a high likelihood of diagnosis of HP. Other features like female sex, mid-inspiratory squeaks, absence of smoking history, and obstructive or mixed defects on PFT were considered to have limited diagnostic utility and regarded as predictors of the disease [8••].
Antigen Identification
The first and foremost step in the approach to diagnose a case of HP is the identification of an offending antigen. Absence of identifiable antigen is associated with worse survival and poor outcomes [31, 32]. Previous studies have used exhaustive questionnaires for the same; however, none has been validated [33, 34]. Antigenic distribution varies with different geographic location, and hence, questionnaires may require to differ depending on the prevalence of various antigens locally. A detailed proforma to elicit occupational and environmental history is paramount, to avoid missing any exposure [28–30]. Tables 1 and 2 enclose case report forms (CRFs) or proforma for potential domestic and occupational exposures.
Antigen-Specific Tests: Serum Antigen–Specific Immunoglobulin Test, Specific Inhalation Challenge Test, and Lymphocyte Proliferation Test
The previously commonly used serum antigen–specific immunoglobulin tests have found a limited place in the recent guidelines. A positive test result only suggests sensitization of the individual to the tested antigen but does not necessitate contribution to the disease unless a positive history of exposure syncing with chronology of symptoms is established [35–37]. There are innumerable antigens, both overt and occult, getting tested for each is practically not possible. Additionally, lack of standardized antigenic preparations and lack of standard techniques with variable diagnostic cutoff thresholds further question their utility. While a low confidence suggestion was made in favor of their use by some [8••], other consensus have not supported their use [7••, 29].
Studies have also used lymphocyte proliferation tests (LPTs) and specific inhalation challenge (SIC) tests in the past [30–34]. They require an antigen extract, the preparation of which lacks standardization. While the SIC test has been deemed to be risky, LPT lacks validated criteria to define a positive response. With the availability of safer and more reliable diagnostic tools, they are no longer recommended as a diagnostic modality in the recent guidelines [7••, 8••, 29].
Radiology
Similar to the 2018 IPF guidelines, patterns on high-resolution computed tomography (HRCT) scan of the chest have been classified into 3 categories for a more confident diagnosis—typical for HP, compatible with HP, and indeterminate for HP. HP affects both lung parenchyma and small airways. A typical HP HRCT pattern essentially requires at least one abnormality indicative of each lung parenchyma and small airway involvement. The described HRCT patterns indicative of parenchymal involvement for typical nfHP are diffuse distribution of ground-glass opacities (GGO) or mosaic attenuation. Small airway disease on imaging may be represented by the presence of ill-defined centrilobular nodules (on inspiratory scans) or air trapping (on expiratory scans) and are common to both fHP and nfHP (Figs. 2 and 3). Subtle GGO, consolidation, or lung cysts distributed diffusely or with peribronchovascular predominance and/or lower lobe predominance attribute to radiology compatible with nfHP. Lung fibrosis coexisting with signs of bronchiolar obstruction suggests fHP. This fibrotic pattern may have a uniform distribution with basal sparing or have predominance in the mid-zone and is indicative of typical fHP. The previously described “head cheese” sign for cHP has been renamed to “three-density pattern.” It indicates a combination of three attenuations on inspiratory CT images, i.e., normal appearing lung, high attenuation (ground-glass opacities), and lucent lung (region of reduced attenuation and vascularity), sharply demarcated from each other. The three-density pattern has been found to be highly specific for fHP and helps differentiate it from IPF. When the pattern or distribution of lung fibrosis deviates from typical, it is categorized as compatible with fHP. The pattern of lung fibrosis may vary from usual interstitial pneumonitis (UIP) to extensive GGOs with subtle fibrotic changes, distributed in subpleural or peribronchovascular pattern (Fig. 4). Upper lobe predominance may help differentiate HP from IPF; however, it has been noted in less than 10% of patients with HP. When radiology shows a UIP pattern or fibrotic non-specific interstitial pneumonia (NSIP) pattern or organizing pneumonia (OP)–like pattern alone, in the absence of other features suggestive of HP, it is classified as indeterminate for fHP [8••].

Fibrotic hypersensitivity pneumonitis: a inspiratory and b expiratory axial HRCT images below the level of tracheal bifurcation show accentuation of air trapping in the expiratory images

Non-fibrotic hypersensitivity pneumonitis: axial HRCT image below the level of tracheal bifurcation shows ill-defined bronchocentric nodules of ground glass attenuation in bilateral lung parenchyma

Fibrotic hypersensitivity pneumonitis: a coronal and b sagittal multiplanar reconstructed images show peribronchovascular fibrosis in the upper lobes of bilateral parenchyma with ill-defined centrilobular nodules in the background
BAL Fluid Analysis
Radiologically, nfHP is often confused with infectious etiologies or sarcoidosis while fHP is commonly confused with IPF. Almost half of patients misdiagnosed with IPF, based on the 2011 criteria, were subsequently diagnosed with chronic HP attributable to identification of an occult antigen with a compatible histopathology [42]. Though above-mentioned modalities help reach a diagnosis in the majority, a small minority of patients may require additional tests. It is imperative to have a confident diagnosis as there are differences in treatment and prognostic implications.
Literature suggests BAL fluid lymphocytosis exists in HP irrespective of its subtype. BAL fluid lymphocytosis was found to be higher in HP than IPF or sarcoidosis, with a proposed cut-off at 30% to distinguish amongst them [8••]. BAL fluid analysis also holds a prognostic value with lymphocytic BAL indicative of steroid responsiveness [44]. Although Perez et al. [7••] did not suggest routine use of BAL in a background clinicoradiological picture compatible with HP, Raghu and coworkers [8••] recommended the same. While the recommendation was made mainly for use in nfHP to identify and exclude pulmonary infections, a favorable weak suggestion was made for fHP [8••]. BAL site should be selected based on the part of the lung most affected, as reflected on the HRCT scan. A minimum of 100 ml and a maximum of 300 ml of normal saline should be instilled in 3–5 sequential aliquots during the procedure. An adequate sample requires a minimum 30% of the instilled saline to be retrieved with suction pressures less than 100 mm Hg. It should be transported to the laboratory within 30 min of procurement, lest it must be transported on ice. Differential cell count analysis requires an optimal volume of 10–20 ml of pooled BAL sample [45].
Lung Biopsy
Specimens for histopathology can be obtained by any of the following techniques: transbronchial lung biopsy (TBLB), transbronchial cryobiopsy (TBLC), and surgical lung biopsy (SLB). The choice amongst the above procedures depends on disease type, patient characteristics, expertise of the clinician, availability of the equipment, potential complication rates, etc. TBLB specimens have a high yield for diagnosis of bronchocentric diseases [35, 46]. It is suggested in cases of suspected nfHP, since granulomas are more likely to be seen and are diagnostic of nfHP. Fibrotic HP specimens obtained through TBLB are small sized, often with crush artifacts that yield non-specific fibrotic pathological features, thus may fail to identify ancillary features differentiating other ILDs especially IPF [8••, 35, 47, 48]. The limitations associated with TBLB were overcome by the evolution of cryo-lung biopsy technique. TBLC specimens are larger with little crush artifacts and feasible for patients unfit to undergo an invasive SLB [49, 50]. Its utility was favored for suspected fHP populations only, to avoid potentially risk-associated SLB [8••]. SLB, though gold standard for histopathological diagnosis, is associated with high complication rates (acute exacerbation of ILD, post-op pneumonia; pleural effusion; chronic chest pain; prolonged air leak; requirement of mechanical ventilation; neuropathic pain) [35, 51–53].
Pathology
Similar to radiological classification, the ATS has classified histopathology of HP into typical, probable, and indeterminate for HP. Typical nfHP requires presence of all of the following features on histopathology: (i) cellular interstitial pneumonia, small lymphocyte predominant, distributed mainly along the bronchioles; (ii) cellular chronic bronchiolitis with cytology similar to interstitial pneumonitis; (iii) small, poorly formed granulomas which may contain cytoplasmic inclusions; and (iv) absence of features suggestive of alternative diagnosis (plasma cell predominance, extensive lymphoid hyperplasia, aspirated particles). When histopathology shows all of the above characteristics except the presence of granulomatous inflammation, it is classified as probable nfHP. Presence of either cellular bronchiolocentric interstitial pneumonia or cellular chronic bronchiolitis is classified as histopathology indeterminate for nfHP [8••].
Typical fHP histopathology contains features suggestive of (i) chronic fibrosing interstitial pneumonia (characterized by architectural distortion, fibroblast foci with or without subpleural honeycombing or fibrotic NSIP-like pattern); OR (ii) airway-centered fibrosis (characterized by peribronchiolar metaplasia or bridging fibrosis); AND (iii) poorly formed non-necrotizing granulomas; AND (iv) absence of alternative histopathology features similar to that of nfHP. Presence of either of the former two criteria, without granulomas, is classified as probable fHP. When histopathology shows features of chronic fibrosing interstitial pneumonia only, it is categorized as indeterminate for fHP. The histopathology pattern of fibrotic HP often overlaps with the “UIP-like pattern.” However concomitant areas of cellular interstitial pneumonia or cellular bronchiolitis or OP-like pattern on biopsy samples help differentiate the two. Biopsy samples thus should be taken from more than one site and more than one lobe to increase the yield of diagnosis [8••].
Treatment
Antigen avoidance lays the foundation of treatment. Studies have documented a mortality benefit in patients with fHP, in whom antigen could be avoided [38]. Studies related to therapeutic interventions are briefly summarized in Table 3. Sadeleer et al. noted significantly improved lung function trends for nfHP, non-significant positive trends for fHP with no survival benefit in either of the cohorts when antigen was avoided. Antigen could not be identified in 20% of their population, which was associated with worse survival [39]. Nishida et al. found nfHP patients experiencing recurrence or progressing to fibrosis with ongoing antigen exposure [54•].
Pharmacological therapies predominantly have two arms—the immunosuppressants and the newly found role of antifibrotics. The immunosuppressant group constitutes the corticosteroids, azathioprine, mycophenolate mofetil (MMF), and rituximab.
Studies on the effect of corticosteroid treatment in HP have been scarce. In a double-blind placebo-controlled trial conducted almost 30 years ago on patients with acute HP, an 8-week course of prednisolone was associated with significantly improved lung functions without a survival benefit [55]. Decades later, the efficacy of corticosteroid was retrospectively studied in a heterogeneous population of HP patients. Corticosteroids were effective in reversing the declining lung functions for the nfHP cohort, without a survival benefit. No significant lung function changes were observed for fHP patients [39]. The role of corticosteroids thus seems restricted to the nfHP subtype. Improved lung function trends point towards improvement in disease progression; however, given no mortality benefit, its associated risks (hyperglycemia, hypertension, osteoporosis, immunocompromised status, aggravation of peptic ulcer disease, weight gain, proximal muscle weakness, cushingoid facies) versus potential benefits must be weighed on a case-to-case basis.
Steroid-related side effects and progressive course of disease despite use have urged upon the need to identify the effect and tolerability of steroid-sparing drugs, viz., MMF, azathioprine, and rituximab. Available data on effects of MMF and azathioprine are limited to retrospective, single-center studies, conducted on a small patient population. Morriset et al. first studied the effect of azathioprine or MMF in patients with chronic HP (cHP). Significantly improved diffusion capacity of lung for carbon monoxide (DLCO) with non-significant increase in forced vital capacity (FVC) was observed. Both drugs were well tolerated and reduced the required dose of steroid, hence the associated side effects [56]. Similar results were documented by Fiddler et al. Compared to Morriset et al., a higher proportion of patients experienced side effects leading to discontinuation of treatment [57]. Adegunsoye et al. studied the outcomes of azathioprine or MMF, added to baseline prednisolone treatment, in patients with cHP. An increased mortality was seen in patients requiring immunosuppressants than those who did not. There was no difference between treatment groups for lung function decline or survival. However, MMF was better tolerated, reduced the required dose of prednisolone, and had the least side effect profile. Hence, use of steroid-sparing immunosuppressants was advocated when deemed necessary [58]. However, all these studies are retrospective analysis of hospital records and case–control datasets. Alexandre et al. observed improved FVC with a stable DLCO, 2 years after treatment with azathioprine alone. Patients who discontinued the therapy were 43.5%, mainly due to disease worsening and liver toxicity [59]. Raimundo et al. assessed the potential factors associated with clinical response in cHP patients treated with azathioprine for 12 months. They were categorized into “responders” and “non-responders” based on FVC decline of > 10% from baseline. BAL lymphocytosis was reported to be significantly higher in responders (36.49% ± 23.25% versus 17.86 ± 15.58), while the “non-responders” had significantly high HRCT findings of traction bronchiectasis (78.3% vs 47.2%) and honeycombing (52.2% vs 25%). BAL lymphocyte count > 28% was found to hold high specificity (88.9%) in identifying the responders, albeit with limited sensitivity (66.7%). Azathioprine was effective in disease stabilization for 78% of their study population [60]. There is limited data on the utility of rituximab in HP with literature limited to a few case reports and case series [61–64]. Rituximab was first used in a treatment refractory case of HP who was concurrently treated with intravenous methylprednisolone. Two months post treatment, subjective and objective improvements were documented which was temporally associated with the use of rituximab [61]. Recently, Ferreira et al. found rituximab to be well tolerated with trend towards disease stabilization or improvement in some patients with cHP [64]. The available evidence seems abstract given the chance factor benefit attributed to the use of rituximab. The side effects of rituximab must be considered, given the high risk of infections, propensity to flare latent tuberculosis and cause allergic reactions.
IPF and other fibrotic ILDs tend to have a similar clinical course and pathogenic mechanisms. Based on this principle, the efficacy of nintedanib in slowing the rate of decline in FVC was examined in the recently concluded INBUILD trial. It was a prospective, randomized, double-blind placebo-controlled trial on patients with progressive fibrosing ILD other than IPF. The cohort comprised 5 subgroups, with cHP contributing to the majority at 26%. Nintedanib given for 52 weeks slowed the rate of FVC decline (− 80.8 ml/year vs − 187.8 ml/year) with its benefits sustained irrespective of the fibrotic pattern on HRCT (95% CI, 70.8 to 185.6; p < 0.001). While it was beneficial in limiting the progress of the disease, there was no significant difference in outcomes like patient mortality, exacerbations, or symptom burden. Adverse event profile was similar in the two groups [65••]. Wells et al. extrapolated the results of the INBUILD trial for each of the 5 subgroups individually. They reported results consistent with those of pilot study across all 5 subgroups with rate of FVC decline in cHP being 73.1 ml/year and a consistent overall safety profile [66•]. In the same study population, Cottin et al. evaluated the potential impact of immunomodulatory therapies on the efficacy and safety of nintedanib. They observed no effect of immunomodulatory therapies on the efficacy of nintedanib. This study, however, did not have a subgroup analysis for each ILD individually [67•].
While INBUILD trial established the role of nintedanib in non IPF-ILDs, the role of pirfenidone was explored in the RELIEF trial. It was a double-blind, randomized, placebo-controlled trial, investigating the efficacy and safety of pirfenidone, conducted on fibrotic ILD patients, with cHP contributing maximally at 45%. The study was prematurely terminated due to slow recruitment. The available data analysis suggested a significantly reduced decline in FVC% predicted and DLCO% predicted in the treatment group. No significant difference was seen with regard to progression-free survival and quality of life (QoL). The adverse events were equally distributed between the two arms. Safety and tolerability profile of pirfenidone was similar to the ones described in IPF trials. Subgroup analysis could not be done due to a small sample size. Although the study was terminated prematurely, the available data favored the use of pirfenidone [68•].
Tzilas et al. analyzed the role of antifibrotics (pirfenidone or nintedanib) in patients with cHP, previously misdiagnosed as IPF. Significantly reduced annual decline in FVC% predicted (4.2%, 95% CI 1.9–6.6%) and DLCO% predicted (5.7%, 95% CI 3.4–8.1%) were observed. The drugs exhibited an acceptable safety profile with 20% patients discontinuing the treatment due to adverse effects [69]. Shibata et al. evaluated the efficacy of pirfenidone in patients with cHP. Significantly improved vital capacity was observed at 6 months [70]. In an open-label study by Mateos et al., efficacy and safety of pirfenidone were studied in patients with cHP. After 1 year of treatment, non-significant improved trends were noted for FVC and DLCO with significantly improved QoL score [71]. All the above-mentioned studies were limited by their methodologies and a small sample size. Drawing conclusions from the same may thus be tricky.
The results of the INBUILD trial provide a hallmark change in the treatment strategy for cHP which was largely limited to antigen avoidance and immunosuppressants. Although nintedanib did not provide a survival benefit, its role in limiting disease progression with a good tolerance and safety profile cannot be underestimated. It thus provides a comforting prospect for fHP patients with limited treatment options, struggling for alternatives to immunosuppressants.
The above-discussed pharmacological therapies suggest a role in slowing the disease progression, that too in a subset of the population with HP. A definitive role in the treatment may thus be served by lung transplant only. Kern et al. reviewed clinical characteristics, survival, and acute or chronic rejection for 31 lung transplant recipients with HP and compared them to 91 referents with IPF. HP subjects had a better medium-term survival after transplant with a reduced risk of death. HP recurred in 2 allografts [72]. Lung transplant thus remains a last resort in the treatment due to a stringent selection criteria, nature of the procedure, risks associated with lifelong immunosuppression, and costs incurred.
Unanswered Questions
Research in the past decade has opened many unknown avenues in HP. However, a multitude of questions still remain unanswered. As HP occurs in genetically susceptible individuals, factors which predispose the patient to the same remain unanswered. Considering significant radiological overlap between IPF and fHP, it would be intriguing to know if more IPF patients actually represent populations with end stage fHP of unknown exposure. The role of antifibrotics in improving outcomes with regard to lung functions and mortality in HP requires further research. It would also be interesting to explore if there exists some radiological or pathological characteristics, predictive of better outcomes with the immunosuppressant group of drugs including azathioprine, mycophenolate mofetil, and rituximab.
Conclusion
We have come a long way today with our understanding and approach to a case of HP. The first official guidelines on the diagnosis of the disease are now available, which have clarified the classification and diagnostic criteria for HP and its phenotypes. A number of trials have further explored the role of immunomodulatory and antifibrotic drugs pertaining to the disease. Although lacunae still exist in our understanding of the factors affecting disease progression and prognosis, we have achieved considerable milestones with our research, the baton for which shall continue to move forward.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Campbell JM. Acute symptoms following work with hay. Br Med J. 1932;11:1143.
Pepys JP, Jenkins PA, Festenstein GN, Lacey M, Gregory PH, Skinner FA. Farmer’s lung thermophilic actinomycetes as a source of “farmer’s lung hay” antigen. The Lancet. 1963;282(7308):607–11.
Richerson HB, Bernstein IL, Fink JN, Hunninghake GW, Novey HS, Reed CE, Salvaggio JE, Schuyler MR, Schwartz HJ, Stechschulte DJ. Guidelines for the clinical evaluation of hypersensitivity pneumonitis: report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84(5):839–44.
Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314–24.
Quirce S, Vandenplas O, Campo P, Cruz MJ, de Blay F, Koschel D, Moscato G, Pala G, Raulf M, Sastre J, Siracusa A. Occupational hypersensitivity pneumonitis: an EAACI position paper. Allergy. 2016;71(6):765–79.
Vasakova M, Morell F, Walsh S, Leslie K, Raghu G. Hypersensitivity pneumonitis: perspectives in diagnosis and management. Am J Respir Crit Care Med. 2017;196(6):680–9.
•• Pérez ER, Travis WD, Lynch DA, Brown KK, Johannson KA, Selman M, Ryu JH, Wells AU, Huang YC, Pereira CA, Scholand MB. Diagnosis and evaluation of hypersensitivity pneumonitis: chest guideline and expert panel report. Chest. 2021 Aug 1;160(2):e97–156. These diagnostic guidelines on hypersensitivity pneumonitis endorsed a multimodality approach and the crux formed by radiology for its diagnosis with limited utility of previously used serological tests.
•• Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, Bargagli E, Chung JH, Collins BF, Bendstrup E, Chami HA. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2020 Aug 1;202(3):e36–69. First diagnostic guidelines on hypersensitivity pneumonitis. They emphasized the core role of radiology in its diagnosis and proposed a new radiological and pathological classification for its two phenotypes.
Thomeer MJ, Costabel U, Rizzato G, Poletti V, Demedts M. Comparison of registries of interstitial lung diseases in three European countries. Eur Respir J. 2001;18(32 suppl):114s-s118.
Kreuter M, Herth FJ, Wacker M, Hammerl P, Wiederhold C, Leidl R, Hellmann A, Pfeifer M, Behr J, Witt S, Kauschka D. Interims analysis of the EXCITING-ILD registry (registry for exploring clinical and epidemiological characteristics of interstitial lung diseases).
Moore I, Wrobel J, Rhodes J, Lin Q, Webster S, Jo H, Troy L, Grainge C, Glaspole I, Corte TJ. Australasian interstitial lung disease registry (AILDR): objectives, design and rationale of a bi-national prospective database. BMC Pulm Med. 2020;20(1):1.
Singh S, Collins BF, Sharma BB, Joshi JM, Talwar D, Katiyar S, Singh N, Ho L, Samaria JK, Bhattacharya P, Gupta R. Interstitial lung disease in India. Results of a prospective registry. Am J Respir Crit Care Med. 2017 Mar 15;195(6):801–13.
Dhooria S, Agarwal R, Sehgal IS, Prasad KT, Garg M, Bal A, Aggarwal AN, Behera D. Spectrum of interstitial lung diseases at a tertiary center in a developing country: a study of 803 subjects. PLoS One. 2018;13(2): e0191938.
Fernández Pérez ER, Kong AM, Raimundo K, Koelsch TL, Kulkarni R, Cole AL. Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: a claims-based cohort analysis. Ann Am Thorac Soc. 2018;15(4):460–9.
Rittig AH, Hilberg O, Ibsen R, Løkke A. Incidence, comorbidity and survival rate of hypersensitivity pneumonitis: a national population-based study. ERJ Open Res. 2019 Oct 1;5(4).
Dhooria S, Sehgal IS, Agarwal R, Muthu V, Prasad KT, Kathirvel S, Garg M, Bal A, Aggarwal AN, Behera D. Incidence, prevalence, and national burden of interstitial lung diseases in India: Estimates from two studies of 3089 subjects. PLoS One. 2022;17(7): e0271665.
Dakhama A, HEGELE RG, Laflamme G, Israel-Assayag E, Cormier Y. Common respiratory viruses in lower airways of patients with acute hypersensitivity pneumonitis. Am J Respir Crit Care Med. 1999 Apr 1;159(4):1316–22.
Hoppin JA, Umbach DM, Kullman GJ, Henneberger PK, London SJ, Alavanja MC, Sandler DP. Pesticides and other agricultural factors associated with self-reported farmer’s lung among farm residents in the Agricultural Health Study. Occup Environ Med. 2007;64(5):334–41.
Singh S, Collins BF, Bairwa M, Joshi JM, Talwar D, Singh N, Samaria JK, Mangal DK, Singh V, Raghu G. Hypersensitivity pneumonitis and its correlation with ambient air pollution in urban India. Eur Respir J. 2019 Feb 1;53(2).
Hanak V, Golbin JM, Ryu JH. Causes and presenting features in 85 consecutive patients with hypersensitivity pneumonitis. In Mayo Clin Proc. 2007 Jul 1;82(7):812–816. Elsevier.
Glazer C, Martyny J, Rose C. Hot tub associated granulomatous lung disease from mycobacterial bioaerosols. Clin Pulm Med. 2008;15(3):138–44.
Gupta A, Rosenman KD. Hypersensitivity pneumonitis due to metal working fluids: sporadic or under reported? Am J Ind Med. 2006;49(6):423–33.
Koschel D, Wittstruck H, Renck T, Müller-Wening D, Höffken G. Presenting features of feather duvet lung. Int Arch Allergy Immunol. 2010;152(3):264–70.
Furuiye M, Miyake S, Miyazaki Y, Ohtani Y, Inase N, Umino T, Yoshizawa Y. Effect of cigarette smoking on the development of murine chronic pigeon breeder’s lung The difference between a short-term and a long-term exposure. J Med Dent Sci. 2007;54(1):87–95.
Sharma BB, Singh S, Singh V. Hypersensitivity pneumonitis: the dug-well lung. In Allergy Asthma Proc 2013 Nov 1;34(6):e59–64.
Møller J, Hyldgaard C, Kronborg-White SB, Rasmussen F, Bendstrup E. Hypersensitivity pneumonitis among wind musicians–an overlooked disease? Eur Clin Respir. 2017;4(1):1351268.
Lacasse Y, Selman M, Costabel U, Dalphin JC, Ando M, Morell F, Erkinjuntti-Pekkanen R, Muller N, Colby TV, Schuyler M, Cormier Y. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952–8.
Morisset J, Johannson KA, Jones KD, Wolters PJ, Collard HR, Walsh SL, Ley B. Identification of diagnostic criteria for chronic hypersensitivity pneumonitis. An International Modified Delphi Survey. Am J Respir Crit Care Med. 2018 Apr 15;197(8):1036–44.
Singh S, Sharma BB, Bairwa M, Gothi D, Desai U, Joshi JM, Talwar D, Singh A, Dhar R, Sharma A, Ahluwalia B. Management of interstitial lung diseases: a consensus statement of the Indian Chest Society (ICS) and National College of Chest Physicians (NCCP). Lung India: Official Organ of Indian Chest Society. 2020;37(4):359.
Suhara K, Miyazaki Y, Okamoto T, Yasui M, Tsuchiya K, Inase N. Utility of immunological tests for bird-related hypersensitivity pneumonitis. Respir Investig. 2015;53(1):13–21.
Masuo M, Miyazaki Y, Suhara K, Ishizuka M, Fujie T, Inase N. Factors associated with positive inhalation provocation test results in subjects suspected of having chronic bird-related hypersensitivity pneumonitis. Respir Investig. 2016;54(6):454–61.
Muñoz X, Sánchez-Ortiz M, Torres F, Villar A, Morell F, Cruz MJ. Diagnostic yield of specific inhalation challenge in hypersensitivity pneumonitis. Eur Respir J. 2014;44(6):1658–65.
Ohtani Y, Kojima K, Sumi Y, Sawada M, Inase N, Miyake S, Yoshizawa Y. Inhalation provocation tests in chronic bird fancier’s lung. Chest. 2000;118(5):1382–9.
Mendoza F, Melendro EI, Baltazares M, Banales JL, Ximenez C, Chapela R, Selman M. Cellular immune response to fractionated avian antigens by peripheral blood mononuclear cells from patients with pigeon breeder’s disease. J Lab Clin Med. 1996;127(1):23–8.
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018 Sep 1;198(5):e44–68.
Johannson KA, Elicker BM, Vittinghoff E, Assayag D, de Boer K, Golden JA, Jones KD, King TE, Koth LL, Lee JS, Ley B. A diagnostic model for chronic hypersensitivity pneumonitis. Thorax. 2016;71(10):951–4.
Baur X, Fischer A, Budnik LT. Spotlight on the diagnosis of extrinsic allergic alveolitis (hypersensitivity pneumonitis). J Occup Med Toxicol. 2015;10(1):1–6.
Pérez ER, Swigris JJ, Forssén AV, Tourin O, Solomon JJ, Huie TJ, Olson AL, Brown KK. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest. 2013;144(5):1644–51.
De Sadeleer LJ, Hermans F, De Dycker E, Yserbyt J, Verschakelen JA, Verbeken EK, Verleden GM, Wuyts WA. Effects of corticosteroid treatment and antigen avoidance in a large hypersensitivity pneumonitis cohort: a single-centre cohort study. J Clin Med. 2018;8(1):14.
Millerick-May ML, Mulks MH, Gerlach J, Flaherty KR, Schmidt SL, Martinez FJ, LeVeque RM, Rosenman KD. Hypersensitivity pneumonitis and antigen identification–an alternate approach. Respir Med. 2016;1(112):97–105.
Cormier Y, Letourneau L, Racine G. Significance of precipitins and asymptomatic lymphocytic alveolitis: a 20-yr follow-up. Eur Respir J. 2004;23(4):523–5.
Cormier Y, Bélanger J, Durand P. Factors influencing the development of serum precipitins to farmer’s lung antigen in Quebec dairy farmers. Thorax. 1985;40(2):138–42.
Morell F, Villar A, Montero MÁ, Muñoz X, Colby TV, Pipvath S, Cruz MJ, Raghu G. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013;1(9):685–94.
De Sadeleer LJ, Hermans F, De Dycker E, Yserbyt J, Verschakelen JA, Verbeken EK, Verleden GM, Verleden SE, Wuyts WA. Impact of BAL lymphocytosis and presence of honeycombing on corticosteroid treatment effect in fibrotic hypersensitivity pneumonitis: a retrospective cohort study. Eur Res J. 2020 Apr 1;55(4).
Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, Drent M, Haslam PL, Kim DS, Nagai S, Rottoli P. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185(9):1004–14.
Poletti V, Chilosi M, Olivieri D. Diagnostic invasive procedures in diffuse infiltrative lung diseases. Respiration. 2004;71(2):107–19.
Mukherjee S, Spiteri M. Transbronchial biopsy and usual interstitial pneumonia: a step forward in disease management. Chest. 2006;130(5):1628–9.
Trahan S, Hanak V, Ryu JH, Myers JL. Role of surgical lung biopsy in separating chronic hypersensitivity pneumonia from usual interstitial pneumonia/idiopathic pulmonary fibrosis*: analysis of 31 biopsies from 15 patients. Chest. 2008;134(1):126–32.
Ganganah O, Guo SL, Chiniah M, Li YS. Efficacy and safety of cryobiopsy versus forceps biopsy for interstitial lung diseases and lung tumors: a systematic review and meta-analysis. Respirology. 2016;21(5):834–41.
Iftikhar IH, Alghothani L, Sardi A, Berkowitz D, Musani AI. Transbronchial lung cryobiopsy and video-assisted thoracoscopic lung biopsy in the diagnosis of diffuse parenchymal lung disease. A meta-analysis of diagnostic test accuracy. Ann Am Thorac Soc. 2017 Jul;14(7):1197–211.
Han Q, Luo Q, Xie JX, Wu LL, Liao LY, Zhang XX, Chen RC. Diagnostic yield and postoperative mortality associated with surgical lung biopsy for evaluation of interstitial lung diseases: a systematic review and meta-analysis. J Thorac Cardiovasc Surg. 2015;149(5):1394–401.
Hutchinson JP, Fogarty AW, McKeever TM, Hubbard RB. In-hospital mortality after surgical lung biopsy for interstitial lung disease in the United States. 2000 to 2011. Am J Respir Crit Care Med. 2016 May 15;193(10):1161–7.
Raj R, Brown KK. Mortality related to surgical lung biopsy in patients with interstitial lung disease. The devil is in the denominator. Am J Respir Crit Care Med. 2016 May 15;193(10):1082–4.
• Nishida T, Kawate E, Ishiguro T, Kanauchi T, Shimizu Y, Takayanagi N. Antigen avoidance and outcome of nonfibrotic and fibrotic hypersensitivity pneumonitis. ERJ Open Res. 2022 Jan 1;8(1). The study highlights the repercussions of persistent antigenic exposure, leading to recurrence of the disease or its progression to fibrosis.
Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer’s lung 1–3. Am Rev Respir Dis. 1992;145:3–5.
Morisset J, Johannson KA, Vittinghoff E, Aravena C, Elicker BM, Jones KD, Fell CD, Manganas H, Dubé BP, Wolters PJ, Collard HR. Use of mycophenolate mofetil or azathioprine for the management of chronic hypersensitivity pneumonitis. Chest. 2017;151(3):619–25.
Fiddler CA, Simler N, Thillai M, Parfrey H. Use of mycophenolate mofetil and azathioprine for the treatment of chronic hypersensitivity pneumonitis—a single-centre experience. Clin Respir J. 2019;13(12):791–4.
Adegunsoye A, Oldham JM, Pérez ER, Hamblin M, Patel N, Tener M, Bhanot D, Robinson L, Bullick S, Chen L, Hsu S. Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ Open Res. 2017 Jul 1;3(3).
Alexandre AT, Martins N, Raimundo S, Melo N, Mota PC, e Bastos HN, Pereira JM, Cunha R, Guimarães S, Moura CS, Morais A. Impact of azathioprine use in chronic hypersensitivity pneumonitis patients. Pulm Pharmacol Ther. 2020 Feb 1;60:101878.
Raimundo S, Pimenta AC, Cruz-Martins N, Rodrigues MC, Melo N, Mota PC, Sokhatska O, Bastos HN, Beltrão M, Guimarães S, Moura CS. Insights on chronic hypersensitivity pneumonitis’ treatment: factors associated with a favourable response to azathioprine. Life Sci. 2021;1(272): 119274.
Lota HK, Keir GJ, Hansell DM, Nicholson AG, Maher TM, Wells AU, Renzoni EA. Novel use of rituximab in hypersensitivity pneumonitis refractory to conventional treatment. Thorax. 2013;68(8):780–1.
Keir GJ, Maher TM, Ming D, Abdullah R, de Lauretis A, Wickremasinghe M, Nicholson AG, Hansell DM, Wells AU, Renzoni EA. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology. 2014;19(3):353–9.
Tamm AM, Kremens K. Rituximab for salvage therapy of refractory hypersensitivity pneumonitis. WMJ. 2019;118(2):95–7.
Ferreira M, Borie R, Crestani B, Rigaud P, Wemeau L, Israel-Biet D, Leroy S, Quétant S, Plantier L, Dalphin JC, Cottin V. Efficacy and safety of rituximab in patients with chronic hypersensitivity pneumonitis (cHP): a retrospective, multicentric, observational study. Respir Med. 2020;1(172): 106146.
•• Flaherty KR, Wells AU, Cottin V, Devaraj A, Inoue Y, Richeldi L, Walsh S, Stowasser S, Coeck C, Goeldner RG, Clerisme-Beaty E. Nintedanib in patients with chronic fibrosing interstitial lung diseases with progressive phenotype: the INBUILD trial. Break-through trial in the management of non-IPF progressive fibrosing ILD. It proved the efficacy of nintedanib in reducing the rate of FVC decline, thus establishing a definitive role in the treatment of patients with progressive fibrosing ILD other than IPF.
• Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, Moua T, Crestani B, Wuyts WA, Stowasser S, Quaresma M. Nintedanib in patients with progressive fibrosing interstitial lung diseases—subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. The Lancet Respiratory Medicine. 2020;8(5):453–60. This subgroup analysis of the INBUILD trial found that the efficacy of nintedanib in reducing the rate of FVC decline was sustained in the sub-population of chronic HP patients.
• Cottin V, Richeldi L, Rosas I, Otaola M, Song JW, Tomassetti S, Wijsenbeek M, Schmitz M, Coeck C, Stowasser S, Schlenker-Herceg R. Nintedanib and immunomodulatory therapies in progressive fibrosing interstitial lung diseases. Respir Res. 2021;22(1):1–9. The study found no effect of commonly used immunomodulatory drugs on the efficacy of nintedanib in reducing the decline of FVC in patients with progressing fibrosing ILD other than IPF.
• Behr J, Prasse A, Kreuter M, Johow J, Rabe KF, Bonella F, Bonnet R, Grohe C, Held M, Wilkens H, Hammerl P. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebo-controlled, phase 2b trial. Lancet. 2021;9(5):476–86. It was the first randomized double blind placebo controlled trial of pirfenidone in non IPF progressive fibrosing ILD. Though prematurely terminated, the methodology and results open avenues for conducting future studies on the role of pirfenidone.
Tzilas V, Tzouvelekis A, Bouros E, Karampitsakos T, Ntassiou M, Avdoula E, Trachalaki A, Antoniou K, Raghu G, Bouros D. Clinical experience with antifibrotics in fibrotic hypersensitivity pneumonitis: a 3-year real-life observational study. ERJ Open Res. 2020 Oct 1;6(4).
Shibata S, Furusawa H, Inase N. Pirfenidone in chronic hypersensitivity pneumonitis: a real-life experience. Sarcoidosis Vasc Diffuse Lung Dis. 2018;35(2):139.
Mateos-Toledo H, Mejía-Ávila M, Rodríguez-Barreto Ó, Mejía-Hurtado JG, Rojas-Serrano J, Estrada A, Castillo-Pedroza J, Castillo-Castillo K, Gaxiola M, Buendía-Roldan I, Selman M. An open-label study with pirfenidone on chronic hypersensitivity pneumonitis. Arch Bronconeumol. 2020;56(3):163–9.
Kern RM, Singer JP, Koth L, Mooney J, Golden J, Hays S, Greenland J, Wolters P, Ghio E, Jones KD, Leard L. Lung transplantation for hypersensitivity pneumonitis. Chest. 2015;147(6):1558–65.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This was a review article. Only HRCT chest images have been shared where the identity of the patients have not been disclosed. Consent was obtained for the same. No animal rights have been violated in this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Interstitial Lung Disease
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ahluwalia, B., Singh, S. New Paradigms in Hypersensitivity Pneumonitis. Curr Pulmonol Rep 11, 116–131 (2022). https://doi.org/10.1007/s13665-022-00295-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13665-022-00295-5