
Abstract
Purpose of Review
This article aims to provide an overview on the diagnosis and management of hypersensitivity pneumonitis (HP). We will focus on the issues surrounding the lack of an international consensus on the diagnosis of HP, and review the existing treatment options for HP.
Recent Findings
There is emerging international consensus that HP should be classified based on clinical, radiologic, and pathologic features and not solely on disease duration. Environmental assessment and antigen avoidance remains the most important step in managing HP. Antifibrotics may soon become a viable option for chronic HP in the near future. Well-designed prospective controlled studies are underway.
Summary
Substantial gaps still remain in our understanding of the diagnosis and management of HP. Further research should focus on establishing internationally accepted diagnostic criteria and clinical practice guidelines to aid the clinician in the challenging treatment of patients with HP.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
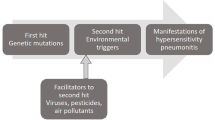
Hypersensitivity pneumonitis (HP), also known as extrinsic allergic alveolitis, is an immune-mediated interstitial lung disease (ILD) triggered by repeated inhalational exposure and sensitization to one or more environmental antigens in genetically susceptible individuals [1••]. Despite our awareness of the disease for decades, substantial gaps remain in our understanding of the pathogenesis, diagnosis, and management of HP. Over the past few years, there has been tremendous interest and a surge of research studies, especially in fibrotic chronic HP (CHP), given its similarities to idiopathic pulmonary fibrosis (IPF), a relentlessly progressive fibrosing lung disease with poor prognosis. In this review article, we highlight the recent developments in our current understanding of HP, in particular CHP, with an emphasis on new research. We also discuss the management of HP with a focus on environmental assessment and antigen avoidance. We conclude with a discussion on the various currently available and future treatment modalities for CHP. Throughout, we also provide our personal insights and observations into the challenging diagnosis and management of HP.
Classification Schema
HP has traditionally been classified as acute, subacute, or chronic based solely on the duration of disease rather than disease features associated with important patient-centered outcomes such as mortality or treatment response [2,3,4,5,6]. Lacasse et al. classified patients with HP into two clusters and suggested that that “subacute HP” may not be a useful term [3]. Building on this, recently proposed classification schema by two multidisciplinary groups of international experts favors categorization of HP into “acute/inflammatory” and “chronic/fibrotic” HP based on “clinical-radiologic-pathologic correlation” [7••, 8••] (Table 1). Acute HP is characterized by predominantly inflammation that is potentially reversible and usually with a symptom duration of less than 6 months. On the contrary, chronic HP is characterized by predominantly irreversible fibrosis, typically with a symptom duration of greater than 6 months, and increased risk of disease progression. This classification schema is borne out of converging evidence in the literature that the presence and extent of fibrosis are associated with increased risk of mortality in HP [9,10,11,12,13]. Although this new proposed classification may provide a more accurate prognosis and inform a better specific treatment strategy [8••], in practice, it may difficult to clearly differentiate the presence and extent of inflammation from fibrosis in a patient with HP as the two may often coexist radiologically and pathologically. This real-world clinical challenge could result in a lost opportunity to reverse a primary inflammatory HP with corticosteroids instead of antifibrotics; conversely, a patient with fibrotic HP treated with corticosteroids and immunosuppression rather than antifibrotics may experience greater potential harm due to drug toxicity in a similar fashion to IPF. Nonetheless, we believe that this novel classification schema of HP is a much-needed, significant step forward towards a consensus diagnostic criterion for HP.
Epidemiology and Etiology
Although limited data on the epidemiology of HP exist, the relatively low prevalence suggests that it is a rare disease. A recent study by Fernandez Perez et al. analyzed insurance claims data in the USA over a decade [14]. They found that the yearly prevalence ranges from 1.67 to 2.71 cases per 100,000 persons and yearly incidence rates ranged from 1.28 to 1.94 cases per 100,000 persons [14]. In a study based on a UK database, the incidence was slightly lower at 0.9 cases per 100,000 persons per year [15]. The prevalence and incidence rates are also likely to differ based on the populations sampled, as well as what is used as the definition of HP. A recent review by Nogueira et al. highlighted six categories of antigens [16]. In terms of clinical utility, the most common clinically important antigens are various molds, organic antigens in the agricultural setting, antigens associated with bird serum, feathers and droppings, and other specific organic and inorganic antigens. As new or previously unrecognized antigens are identified, there will remain a perpetually growing list of HP-associated antigens and sources of exposure [7••].
Pathogenesis
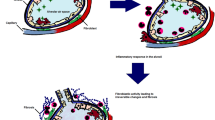
HP is an immunologic reaction to inhaled antigens leading to lung inflammation and possibly fibrosis, mainly involving the distal bronchus and upper lung lobes [17, 18]. A recent review by Vasakova et al. suggested that genetically susceptible individuals develop HP after being exposed to one or more environmental antigens, followed by antigen presentation via the MHC I and MHC II pathways activating CD8+ and CD4+ T lymphocytes, which lead to both cellular and humoral immune responses causing small airway inflammation and granuloma formation, with some patients ultimately developing chronic HP due to fibroproliferation [1••]. Further research is needed to identify and validate the gene polymorphisms that may increase susceptibility to HP [1••].
Clinical Presentation
Patients with acute/inflammatory HP typically develop an acute onset of symptoms within the first few hours to days after being exposed to antigens. Symptoms may be episodic; include cough, dyspnea, wheezing, malaise, fever, and/or chills; and may last for hours to weeks [3]. This syndrome may be misdiagnosed as a viral respiratory infection or community-acquired pneumonia. With repeated exposure, some patients may present with the above symptoms but with differing severity. If not recognized and treated appropriately, some patients, usually after 6 months, experience disease progression to “chronic/fibrotic HP.” Symptoms are similar to other ILDs, most notably dry cough and dyspnea, and increased risk of unpredictable but deadly acute exacerbations.
Diagnostic Evaluation
The first step in diagnosing HP is a comprehensive, thorough patient history with a focus on environmental and occupational exposures. The use of an exposure questionnaire may facilitate history taking and is commonly used at ILD centers, although their clinical utility remains unvalidated. Serological studies, HRCT, bronchoalveolar lavage, and lung biopsy may be needed based on the clinical picture. All of these data may then be reviewed by a multidisciplinary group of pulmonologists, radiologists, and pathologists, ideally with expertise in ILD, for a consensus diagnosis.
Comprehensive History, Physical Examination, and Pulmonary Physiologic Testing
Similar to the approach of any patient with suspected ILD, a comprehensive and thorough patient history must always take place. In addition to ruling out other potential causes of ILD such as connective tissue disease and drug toxicity, the clinician should perform a careful occupational, domestic, avocational, and lifestyle history. Any identified potentially significant exposures should be followed up with additional details such as exposure onset, duration, and intensity. This information would then need to be correlated with symptom and disease onset. In our experience, ILD exposure questionnaires can assist in eliciting history of a potentially significant exposure; however, these questionnaires are unstandardized and their clinical utility remains unvalidated. In addition to a comprehensive history, physical examination is essential to confirm bilateral rales on auscultation, similar to other ILDs, and occasionally inspiratory squeaks [19]. Pulmonary physiologic testing with pulmonary function testing (PFT) and 6-min walk testing should be performed initially for baseline severity and longitudinally for disease monitoring.
Serology
The role of serum-specific IgG (SsIgG) antibodies in the diagnosis of HP remains controversial due to their reported poor sensitivity and specificity [20]. While the presence of precipitating antibodies could confirm clinical suspicion based on history taking, the presence of precipitating antibodies may prompt the clinician to re-examine potentially missed antigens during the initial evaluation. Most panels utilize ELISA, but immunodiffusion and ImmunoCAP may also be used [21]. The presence of SsIgG antibodies simply denotes exposure to the antigens being tested. Importantly, their presence is not diagnostic of HP. The threshold for abnormal levels of IgG against antigens is determined based on studies such as the one by Raulf et al., which determined the cutoff of precipitating antibody levels based on healthy volunteers [22]. In a study by Fenoglio et al., assuming a prevalence of HP of 35% among those tested for HP panel (which is quite high), with “double diffusion” and “electrosyneresis” (different from the commonly used ELISA and immunodiffusion methods available clinically), the sensitivity of said immunologic tests against mold antigens was 76%, with a specificity of 82%, PPV of 69.5%, and NPV of 86.4%. [20].
High-Resolution CT
High-resolution CT of the chest is a key element in the diagnostic evaluation of HP. Studies have shown that a confident radiologic diagnosis of HP can be attained with up to 92% accuracy but only 61% sensitivity [23, 24]. In acute/inflammatory HP, typical radiologic features include symmetric, bilateral, usually upper lobe predominant, ground glass opacities, along with centrilobular nodules and mosaic attenuation [25,26,27]. In chronic/fibrotic HP, fibrotic changes such as reticulation, traction bronchiectasis, and honeycombing can be observed [28]. Mosaic attenuation can be seen in both acute and chronic HP [29]. Interestingly, mosaic attenuation may also be seen in up to 51% of patients with IPF. Therefore, its presence alone is not diagnostic of HP [30•]. The “headcheese” sign may be highly specific (0.93) and moderately sensitive (0.93) for a high confidence diagnosis of fibrotic HP [30•]. In end-stage fibrotic HP, the CT findings may not be distinguishable from IPF with a UIP-like or fibrotic NSIP-like pattern [31].
Bronchoalveolar Lavage
Bronchoalveolar lavage (BAL) should be considered an important diagnostic tool for HP. In addition to ruling out a lower respiratory tract infection and malignancy, BAL fluid cellular analyses may be performed to evaluate the presence of significant lymphocytosis. Various levels of BAL lymphocytosis have been quoted [32], but we typically use a threshold of greater than 30% as suggestive of BAL lymphocytosis. However, according to the Delphi survey study by Morisset et al., BAL lymphocytosis greater than 40% was determined by expert consensus to be regarded as significant for BAL lymphocytosis [33•]. The 2012 ATS guidelines on the use of BAL in ILD suggested a cutoff of 50% for BAL lymphocytosis and recommend against routine use of the CD4+: CD8+ ratio in the diagnosis of HP [34]. Of note, BAL fluid lymphocyte counts may be normal, or lower than normal, in some patients with CHP [32, 33•, 34]. In addition, significant BAL lymphocytosis has also been reported in sarcoidosis, organizing pneumonia, and non-specific interstitial pneumonia [35•].
Lung Biopsy
Transbronchial lung biopsy and surgical lung biopsy are commonly employed methods of obtaining lung tissue samples to aid in the diagnosis of HP. The yield for transbronchial biopsy is low, and the highest reported study showed a yield rate of 40% [36]. Surgical lung biopsy may be necessary in some cases to establish a diagnosis [37]; however, risks of the procedure, especially taking into account the patient’s disease severity, must be carefully weighed against the benefits of a confirmed diagnosis. There is limited evidence for the use of transbronchial cryobiopsy (TBCB) specifically in hypersensitivity pneumonitis. However, the evidence for the use of TBLC in the setting of interstitial lung disease (including HP) showed that the yield rate could be up to 84.4% at a cost of increased risk of bleeding and pneumothorax [38].
Histopathology
In acute HP, characteristic histopathologic features include a classic triad of bronchiolocentric lymphoplasmacytic interstitial infiltrate, chronic bronchiolitis, and poorly formed non-necrotizing granulomas in peribronchial areas [39]. Over time with prolonged exposure, histopathologic fibrosis predominates with UIP-like, fibrotic NSIP-like, or bronchiolocentric patterns [11, 40, 41]. Importantly, the small airways are rarely normal in CHP unlike in UIP of IPF [11, 39].
Diagnostic Criteria
At present, there are no expert consensus and clinical practice guidelines regarding the diagnosis of HP. Furthermore, as demonstrated by the study by Walsh et al., there is low inter-observer agreement by a multidisciplinary team meeting for hypersensitivity pneumonitis (kappa = 0.24), as opposed to a higher agreement for the diagnosis of IPF (kappa = 0.60) [42]. This likely reflects the lack of established diagnostic criteria for HP. In addition, a study by Morell et al. showed that 20 of 46 patients (43%) diagnosed with IPF were subsequently diagnosed with HP after repeatedly questioning the patients for antigen exposure every 4 months [43]. Significant efforts towards development of a more uniform diagnostic criterion continue. Ryerson et al. recently proposed a new standardized diagnostic ontology for fibrotic ILD, namely “confident diagnosis” (90% or above), “provisional high confidence” (70–89%), “provisional low confidence” (51–69%), and “unclassifiable ILD” (50% or less) [44•]. Building on this model, Morisset et al. used multiple rounds of Delphi survey to narrow down the diagnostic features of chronic HP by consensus based on 45 HP experts [33•]. This study suggested 2 clinical scenarios that would warrant a “confident diagnosis” (90% or above) of chronic HP as mentioned above. The first is a combination of identified antigen on history, HRCT features suggestive of chronic HP, and BAL lymphocytosis of greater than 40%. In this scenario, the expert consensus was that a surgical lung biopsy is not necessary. The second scenario where a confident diagnosis could be made would be one that includes an identified antigen, as well as a surgical lung biopsy consistent with chronic HP, regardless of HRCT or BAL cell count findings. Furthermore, this study also listed multiple clinical, radiologic, BAL, and histopathologic features that were felt to meet the “important” threshold for them to be classified as diagnostic features of HP. Although there have been significant efforts in attempting to come up with a consensus diagnostic criteria for chronic HP, there still remains a lack of consensus criteria for the diagnosis of acute HP. Nonetheless, multidisciplinary team meeting of pulmonologists, radiologists, and pathologists, ideally with ILD expertise [,] is generally regarded as an important step in [accurately] diagnosing HP, especially chronic HP when there are many clinical mimickers [45].
Management of HP
Antigen Avoidance
Antigen avoidance is one of the most important aspects of HP management. Identification and removal of an inciting antigen have been shown to improve survival for HP [2], as environmental levels of antigens (such as avian antigens) measured after diagnosis correlate with prognosis [46]. In one study, patients in contact with birds for more than 2 years compared with those with contact for less than 2 years had worse clinical outcomes [47].
A thorough environmental evaluation of the patient’s environment, especially where they spend a considerable amount of time, should be done. This includes the consideration of home, work [48], hobbies, and potential exposures amongst family or friends. At our center, we often recommend a professional home or worksite visit by a certified consultant of the American Industrial Hygienists Association (AIHA) with the aim of antigen identification, especially molds, and subsequent targeted environmental remediation.
There is no proven standardized protocol of environmental assessment or methods to remove antigens from the surrounding. Bulk sampling from the environment and then testing the patients against the antigens extracted have been proposed [49]. If the antigen in question is obvious, such as down feather blanket or pillows, they should be immediately removed. Pets, especially birds, should also be promptly removed. However, one study showed that bird antigen persisted in the home environment even after bird removal, with antigens persisting at high levels up to 18 months [50].
In some cases, when antigen identification and/or avoidance remain impossible with the current home or work situations, a complete change of environment (moving to a new home, switching to a new job) may be necessary, which can potentially cause great emotional and financial consequences for the patient (especially in the agricultural setting).
Immunosuppressive Therapy
In up to 20–30% of patients, the diagnosis of HP may be established without any identifiable antigens [51]. Furthermore, there are some cases when complete antigen avoidance is not possible. At our center, we consider aggressive immunosuppressive therapy for CHP patients with symptomatic and progressive disease (Table 2).
Corticosteroid
The most studied immunosuppressive therapy in HP is corticosteroids. There is one double-blind placebo-controlled study involving patients with HP. Thirty-six patients with farmer’s lung (during the acute stage) were included in this study. Twenty patients were given prednisolone while the rest received a placebo for 8 weeks. There were no differences in FVC, FEV1, or DLCO, or relapse of farmer’s lung in 5 years, but at 1 month, the DLCO was higher in the treatment group [52]. A retrospective study in the 1980s showed that corticosteroid improved chest x-ray findings but had no significant clinical effects in the long term (average follow-up of 18.6 months) [53]. In a more recent retrospective study involving 93 non-fibrotic HP (79% treated with steroids) and 109 fibrotic HP patients (80% treated with steroids), corticosteroid treatment did not result in improved survival or DLCO. However, corticosteroid treatment in non-fibrotic HP patients did improve FVC [54].
Prior to initiating corticosteroids, it has been suggested by some authors to identify whether inflammatory features are present or not [8••]. This approach aims to inform the duration of corticosteroid treatment. Inflammatory features as suggested with this protocol include ground-glass opacities on HRCT, BAL lymphocytosis, or histopathological cellular interstitial pneumonia or granulomatous pattern. The authors suggested that in patients without inflammatory features, the course of corticosteroid should be much shorter. While this strategy has not been prospectively verified, we also agree with this in our clinical practice. We recommend a starting dosage of 0.5–1 mg per kg of prednisone for at least 4 weeks. Then, based on FVC, FEV1, and DLCO, as well as subsequent CT findings, further tapering should be accomplished over the next month down to 20 mg daily of prednisone. Further tapering would require stability of PFTs and CT findings. If the patient has pre-dominant fibrotic features of HP and has an inadequate response after at least 4 weeks of 0.5–1 mg per kg of prednisone, we recommend tapering prednisone off over 2–4 weeks.
Other Immunosuppressive Agents
Other immunosuppressive agents had also been studied, but the most studied are mycophenolate mofetil (MMF) and azathioprine (AZA). A study by Morisset et al. looked at 70 patients and 51 were treated with MMF and 19 with AZA. DLCO improved by a median of 4.2% but not FVC [55]. Adegunsoye et al. studied adding either MMF or AZA to corticosteroids, and discovered that adding MMF or AZA reduced treatment-emergent adverse events, but there were no significant differences between survival and lung function decline [56]. Rituximab was studied in multiple small studies. For example, in a retrospective study of ILD patients treated with rituximab, 6 out of 50 had chronic HP and did show improvement in FVC and DLCO but the small size of this study limits generalizability of this approach [57].
Other Therapies
Given the similarities between progressive fibrotic CHP and IPF [58], there is considerable interest in evaluating the safety and efficacy of the antifibrotics, pirfenidone, and nintedanib, in CHP.
Pirfenidone, an antifibrotic approved for the treatment of patients with IPF, has been evaluated in uncontrolled studies for the treatment of HP as well. A recent study by Shibata et al. studied the use of pirfenidone in chronic HP in 23 patients retrospectively [59]. The decline in vital capacity in the 6 months following therapy was slower compared with the decline in vital capacity in the 6 months prior to the initiation of therapy. No other clinical parameters were reported, and the small size and retrospective nature of this study limit the recommendation of pirfenidone in HP unless further studies are published. Larger, multicenter, randomized controlled trials are actively investigating the use of antifibrotics in non-IPF interstitial lung disease, including chronic HP, such as the RELIEF trial for pirfenidone [60] and the INBUILD trial for nintedanib [61].
If HP continues to progress despite the above treatment, lung transplantation should be considered as a life-saving option. In a study by Kern et al., the clinical outcomes of 31 patients with HP who underwent lung transplantation were compared with 91 patients with IPF who also underwent transplantation. The authors found that patients with HP had a higher survival rate up to 5 years post-op. Furthermore, only 2 out of 31 patients with HP developed recurrent HP in their allografts. These findings suggest that patients with chronic HP do well after lung transplantation [62].
Prognosis
The prognosis of HP is associated with various clinical, laboratory, radiologic, and histopathologic findings. Even after adjustment for the presence of fibrosis, Fernandez Perez et al. found that inability to identify an inciting antigen was independently associated with increased mortality (median, 4.88 years vs. 8.75 years) [2]. One study demonstrated that clubbing is associated with clinical deterioration [63]. PFT findings (decline in FVC > 10% [64], low TLC, and low DLCO [65]) were also found to be associated with prognosis. A clinical prediction model, the ILD-GAP model, was also found to predict mortality in patients with HP [66]. In terms of radiologic findings, as previously mentioned, studies have shown that the presence of fibrosis [67, 68] is characterized by traction bronchiectasis and honeycombing [42]. On the other hand, air trapping and mosaic attenuation are associated with better prognosis [29]. A recent study by Salisbury et al. categorized CHP patients into three CT phenotypes, namely “honeycomb present,” “non-honeycomb fibrosis,” and “non-fibrotic,” and found that patients with honeycombing had the worst prognosis [69•]. In terms of pathologic patterns on lung biopsy, a UIP pattern may suggest worse prognosis [70]. Other tests, not routinely used in clinical practice, such as serum periostin [71], are also associated with worse prognosis in HP.
Conclusion
HP remains a disease that is incompletely understood. International consensus or clinical practice guidelines on how to classify and diagnose HP are non-existent. However, recent studies and expert opinions have suggested that the term “subacute HP” should not be used anymore. Rather, the terms “acute/inflammatory” and “chronic/fibrotic” have greater prognostic and potential therapeutic implications. Major steps have been taken recently to standardize the diagnosis of chronic HP but similar steps have not been made for acute HP. Identifying the inciting antigen(s) is essential. SsIgG by itself is of limited use, but may provide clues to identify the antigen. Other important diagnostic tests include HRCT, BAL cellular analysis, and histopathologic data, if available.
In terms of managing HP, there remains no standardized accepted method of antigen identification and avoidance. Immunosuppressive therapy is recommended when antigen avoidance is impossible or ineffective. The first-line agent is corticosteroid treatment; however, the body of evidence supporting corticosteroid is weak. The duration of steroid should be guided by whether inflammatory features are present or not. MMF, AZA, and rituximab are second-line agents with even more limited evidence. Antifibrotics are currently being investigated in randomized controlled trials in patients with fibrotic HP. Lung transplantation is an option for end-stage fibrotic HP and patients generally do well post-transplant.
Future research in HP should focus on international consensus on classification and diagnosis, as well as prospective studies on the effectiveness of different therapeutic options, especially antifibrotics, based on standardized diagnostic criteria.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Vasakova M, Selman M, Morell F, Sterclova M, Molina-Molina M, Raghu G. Hypersensitivity pneumonitis: current concepts of pathogenesis and potential targets for treatment. Am J Respir Crit Care Medicine. 2019;200:301–8. Important review on our current understanding of HP pathogenesis and potential therapeutic targets for the treatment of HP.
Pérez ER, Swigris JJ, Forssén AV, Tourin O, Solomon JJ, Huie TJ, et al. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest. 2013;144(5):1644–51.
Lacasse Y, Selman M, Costabel U, Dalphin JC, Morell F, Erkinjuntti-Pekkanen R, et al. Classification of hypersensitivity pneumonitis. International archives of allergy and immunology. 2009;149(2):161–6.
Zacharisen MC, Schlueter DP, Kurup VP, Fink JN. The long-term outcome in acute, subacute, and chronic forms of pigeon breeder’s disease hypersensitivity pneumonitis. Annals of Allergy, Asthma & Immunology. 2002;88(2):175–82.
Ohtani Y, Saiki S, Sumi Y, Inase N, Miyake S, Costabel U, et al. Clinical features of recurrent and insidious chronic bird fancier's lung. Annals of Allergy, Asthma & Immunology. 2003;90(6):604–10.
Tateishi T, Ohtani Y, Takemura T, Akashi T, Miyazaki Y, Inase N, et al. Serial high-resolution computed tomography findings of acute and chronic hypersensitivity pneumonitis induced by avian antigen. Journal of computer assisted tomography. 2011;35(2):272–9.
•• Vasakova M, Morell F, Walsh S, Leslie K, Raghu G. Hypersensitivity pneumonitis: perspectives in diagnosis and management. American journal of respiratory and critical care medicine. 2017;196(6):680–9. Key review on HP that proposed classification schema for HP as “acute/inflammatory” vs. “chronic/fibrotic”.
•• Salisbury ML, Myers JL, Belloli EA, Kazerooni EA, Martinez FJ, Flaherty KR. Diagnosis and treatment of fibrotic hypersensitivity pneumonia. Where we stand and where we need to go. American journal of respiratory and critical care medicine. 2017;196(6):690–9. Key review on HP. Recommends treating HP according to whether inflammatory features are present or not.
Pérez-Padilla R, Salas J, Chapela R, Sánchez M, Carrillo G, Pérez R, et al. Mortality in Mexican patients with chronic pigeon breeder’s lung compared with those with usual interstitial pneumonia. American Review of Respiratory Disease. 1993;148:49.
Ohtani Y, Saiki S, Kitaichi M, Usui Y, Inase N, Costabel U, et al. Chronic bird fancier’s lung: histopathological and clinical correlation. An application of the 2002 ATS/ERS consensus classification of the idiopathic interstitial pneumonias. Thorax. 2005;60(8):665–71.
Churg A, Muller NL, Flint J, Wright JL. Chronic hypersensitivity pneumonitis. The American journal of surgical pathology. 2006;30(2):201–8.
Chiba S, Tsuchiya K, Akashi T, Ishizuka M, Okamoto T, Furusawa H, et al. Chronic hypersensitivity pneumonitis with a usual interstitial pneumonia-like pattern: correlation between histopathologic and clinical findings. Chest. 2016;149(6):1473–81.
Vourlekis JS, Schwarz MI, Cherniack RM, Curran-Everett D, Cool CD, Tuder RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. The American journal of medicine. 2004;116(10):662–8.
Pérez F, Evans R, et al. Epidemiology of hypersensitivity pneumonitis among an insured population in the United States: a claims-based cohort analysis. Annals of the American Thoracic Society. 2018;15.4:460–9.
Solaymani-Dodaran M, West J, Smith C, Hubbard R. Extrinsic allergic alveolitis: incidence and mortality in the general population. QJM: An International Journal of Medicine. 2007;100(4):233–7. https://doi.org/10.1093/qjmed/hcm008.
Nogueira R, Melo N, e Bastos HN, Martins N, Delgado L, Morais A, et al. Hypersensitivity pneumonitis: antigen diversity and disease implications. Pulmonology. 2019;25(2):97–108.
Girard M, Lacasse Y, Cormier Y. Hypersensitivity pneumonitis. Allergy. 2009;64:322–34. https://doi.org/10.1111/j.1398-9995.2009.01949.x.
Bourke SJ, Dalphin JC, Boyd G, McSharry C, Baldwin CI, Calvert JE. Hypersensitivity pneumonitis: current concepts. European Respiratory Journal. 2001;18(32 suppl):81s–92s.
Reich JM. Chirping rales in bird-fancier’s lung. Chest. 1993;104(1):326–7.
Fenoglio CM, Reboux G, Sudre B, Mercier M, Roussel S, Cordier JF, et al. Diagnostic value of serum precipitins to mould antigens in active hypersensitivity pneumonitis. European Respiratory Journal. 2007;29(4):706–12.
Van Hoeyveld E, Dupont L, Bossuyt X. Quantification of IgG antibodies to Aspergillus fumigatus and pigeon antigens by ImmunoCAP technology: an alternative to the precipitation technique? Clinical chemistry. 2006;52(9):1785–93.
Raulf M, Joest M, Sander I, Hoffmeyer F, Nowak D, Ochmann U, Preisser A, Schreiber J, Sennekamp J, Koschel D. Update of reference values for IgG antibodies against typical antigens of hypersensitivity pneumonitis. Allergo J Int. 2019;1–2.
Lynch DA, Newell JD, Logan PM, King TE Jr, Müller NL. Can CT distinguish hypersensitivity pneumonitis from idiopathic pulmonary fibrosis?. AJR. American journal of roentgenology. 1995;165(4):807–11.
Silva CI, Muller NL, Lynch DA, Curran-Everett D, Brown KK, Lee KS, et al. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology. 2008;246(1):288–97.
Hansell DM, Wells AU, Padley SP, Müller NL. Hypersensitivity pneumonitis: correlation of individual CT patterns with functional abnormalities. Radiology. 1996;199(1):123–8.
Lynch DA, Rose CS. Way D, King Jr TE. Hypersensitivity pneumonitis: sensitivity of high-resolution CT in a population-based study. AJR. American journal of roentgenology. 1992;159(3):469–72.
Silva CI, Churg A, Müller NL. Hypersensitivity pneumonitis: spectrum of high-resolution CT and pathologic findings. American Journal of Roentgenology. 2007;188(2):334–44.
Sahin H, Brown KK, Curran-Everett D, Hale V, Cool CD, Vourlekis JS, et al. Chronic hypersensitivity pneumonitis: CT features—comparison with pathologic evidence of fibrosis and survival. Radiology. 2007;244(2):591–8.
Chung JH, Zhan X, Cao M, Koelsch TL, Manjarres DC, Brown KK, et al. Presence of air trapping and mosaic attenuation on chest computed tomography predicts survival in chronic hypersensitivity pneumonitis. Annals of the American Thoracic Society. 2017;14(10):1533–8.
• Barnett J, Molyneaux PL, Rawal B, Abdullah R, Hare SS, Vancheeswaran R, et al. Variable utility of mosaic attenuation to distinguish fibrotic hypersensitivity pneumonitis from idiopathic pulmonary fibrosis. European Respiratory Journal. 2019;54(1):1900531. Interesting study that noted the presence of mosaic attenuation in 51% of IPF patients and found the “headcheese sign” to be a highly specific radiologic finding for fibrotic HP.
Aburto M, Herráez I, Iturbe D, Jiménez-Romero A. Diagnosis of idiopathic pulmonary fibrosis: differential diagnosis. Medical Sciences. 2018;6(3):73.
Ohshimo S, Bonella F, Cui A, Beume M, Kohno N, Guzman J, et al. Significance of bronchoalveolar lavage for the diagnosis of idiopathic pulmonary fibrosis. American journal of respiratory and critical care medicine. 2009;179(11):1043–7.
• Morisset J, Johannson KA, Jones KD, Wolters PJ, Collard HR, Walsh SL, et al. Identification of diagnostic criteria for chronic hypersensitivity pneumonitis. An International Modified Delphi Survey. American journal of respiratory and critical care medicine. 2018;197(8):1036–44. Recent Delphi survey among international HP experts on consensus diagnostic features of chronic HP.
Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. American journal of respiratory and critical care medicine. 2012;185(9):1004–14.
• Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. American journal of respiratory and critical care medicine. 2018;198(5):e44–68. Most recent IPF clinical practice guidelines for the diagnosis of IPF.
Adams TN, Newton CA, Batra K, Abu-Hijleh M, Barbera T, Torrealba J, et al. Utility of bronchoalveolar lavage and transbronchial biopsy in patients with hypersensitivity pneumonitis. Lung. 2018;196(5):617–22.
Trahan S, Hanak V, Ryu JH, Myers JL. Role of surgical lung biopsy in separating chronic hypersensitivity pneumonia from usual interstitial pneumonia/idiopathic pulmonary fibrosis*: analysis of 31 biopsies from 15 patients. Chest. 2008;134(1):126–32.
Sharp C, McCabe M, Adamali H, Medford AR. Use of transbronchial cryobiopsy in the diagnosis of interstitial lung disease—a systematic review and cost analysis. QJM: An International Journal of Medicine. 2016;110(4):207–14.
Takemura T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y. Pathology of hypersensitivity pneumonitis. Curr Opin Pulm Med. 2008;14(5):440–54.
Churg A, Sin DD, Everett D, Brown K, Cool C. Pathologic patterns and survival in chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2009;33:1765–70.
Ohtani Y, Kojima K, Sumi Y, Sawada M, Inase N, Miyake S, et al. Inhalation provocation tests in chronic bird fancier’s lung. Chest. 2000;118:1382–9.
Walsh SL, Sverzellati N, Devaraj A, Wells AU, Hansell DM. Chronic hypersensitivity pneumonitis: high resolution computed tomography patterns and pulmonary function indices as prognostic determinants. European radiology. 2012;22(8):1672–9.
Morell F, Villar A, Montero MÁ, Muñoz X, Colby TV, Pipvath S, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. The lancet Respiratory medicine. 2013;1(9):685–94.
• Ryerson CJ, Corte TJ, Lee JS, Richeldi L, Walsh SL, Myers JL, et al. A standardized diagnostic ontology for fibrotic interstitial lung disease. An international working group perspective. American journal of respiratory and critical care medicine. 2017;196(10):1249–54. Provides a framework for the standardization of diagnostic confidence in ILD.
Elicker BM, Jones KD, Henry TS, Collard HR. Multidisciplinary approach to hypersensitivity pneumonitis. Journal of thoracic imaging. 2016;31(2):92–103.
Sema M, Miyazaki Y, Tsutsui T, Tomita M, Eishi Y, Inase N. Environmental levels of avian antigen are relevant to the progression of chronic hypersensitivity pneumonitis during antigen avoidance. Immunity, inflammation and disease. 2018;6(1):154–62.
De Gracia J, Morell F, Bofill JM, Curull V, Orriols R. Time of exposure as a prognostic factor in avian hypersensitivity pneumonitis. Respiratory medicine. 1989;83(2):139–43.
Quirce S, Vandenplas O, Campo P, Cruz MJ, de Blay F, Koschel D, et al. Occupational hypersensitivity pneumonitis: an EAACI position paper. Allergy. 2016;71(6):765–79.
Millerick-May ML, Mulks MH, Gerlach J, Flaherty KR, Schmidt SL, Martinez FJ, et al. Hypersensitivity pneumonitis and antigen identification–an alternate approach. Respiratory medicine. 2016;112:97–105.
Craig TJ, Hershey J, Engler RJ, Davis W, Carpenter GB, Salata K. Bird antigen persistence in the home environment after removal of the bird. Annals of allergy. 1992;69(6):510–2.
Ohshimo S, Bonella F, Guzman J, Costabel U. Hypersensitivity pneumonitis. Immunology and Allergy Clinics. 2012;32(4):537–56.
Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonary function in farmer’s lung 1-3. Am Rev Respir Dis. 1992;145:3–5.
Mönkäre S. Influence of corticosteroid treatment on the course of farmer’s lung. European journal of respiratory diseases. 1983;64(4):283–93.
De Sadeleer L, Hermans F, De Dycker E, Yserbyt J, Verschakelen J, Verbeken E, et al. Effects of corticosteroid treatment and antigen avoidance in a large hypersensitivity pneumonitis cohort: a single-centre cohort study. Journal of clinical medicine. 2019;8(1):14.
Morisset J, Johannson KA, Vittinghoff E, Aravena C, Elicker BM, Jones KD, et al. Use of mycophenolate mofetil or azathioprine for the management of chronic hypersensitivity pneumonitis. Chest. 2017;151(3):619–25.
Adegunsoye A, Oldham JM, Pérez ER, Hamblin M, Patel N, Tener M, et al. Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ open research. 2017;3(3):00016–2017.
Keir GJ, Maher TM, Ming D, Abdullah R, de Lauretis A, Wickremasinghe M, et al. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology. 2014 Apr;19(3):353–9.
Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27:180076.
Shibata S, Furusawa H, Inase N. Pirfenidone in chronic hypersensitivity pneumonitis: a real-life experience. Sarcoidosis vasculitis and diffuse lung disease. 2018;35(2):139–42.
Behr J, Neuser P, Prasse A, Kreuter M, Rabe K, Schade-Brittinger C, et al. Exploring efficacy and safety of oral pirfenidone for progressive, non-IPF lung fibrosis (RELIEF): a randomized, double-blind, placebo-controlled, parallel group, multi-center, phase II trial. BMC Pulm Med. 2017;17:122.
Flaherty KR, Brown KK, Wells AU, Clerisme-Beaty E, Collard HR, Cottin V, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ open respiratory research. 2017;4(1):e000212.
Kern RM, Singer JP, Koth L, Mooney J, Golden J, Hays S, et al. Lung transplantation for hypersensitivity pneumonitis. Chest. 2015;147(6):1558–65.
Sansores R, Salas J, Chapela R, Barquin N, Selman M. Clubbing in hypersensitivity pneumonitis: its prevalence and possible prognostic role. Archives of internal medicine. 1990;150(9):1849–51.
Gimenez A, Storrer K, Kuranishi L, Soares MR, Ferreira RG, Pereira CA. Change in FVC and survival in chronic fibrotic hypersensitivity pneumonitis. Thorax. 2018;73(4):391–2.
Miyazaki Y, Tateishi T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y. Clinical predictors and histologic appearance of acute exacerbations in chronic hypersensitivity pneumonitis. Chest. 2008;134(6):1265–70.
Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014;145(4):723–8.
Mooney JJ, Elicker BM, Urbania TH, Agarwal MR, Ryerson CJ, Nguyen ML, et al. Radiographic fibrosis score predicts survival in hypersensitivity pneumonitis. Chest. 2013;144(2):586–92.
Hanak V, Golbin JM, Hartman TE, Ryu JH. High-resolution CT findings of parenchymal fibrosis correlate with prognosis in hypersensitivity pneumonitis. Chest. 2008;134(1):133–8.
• Salisbury ML, Gu T, Murray S, Gross BH, Chughtai A, Sayyouh M, et al. Hypersensitivity pneumonitis: radiologic phenotypes are associated with distinct survival time and pulmonary function trajectory. Chest. 2019;155(4):699–711. Recent paper on classifying HRCT findings of HP patients into three phenotypes.
Wang P, Jones KD, Urisman A, Elicker BM, Urbania T, Johannson KA, et al. Pathologic findings and prognosis in a large prospective cohort of chronic hypersensitivity pneumonitis. Chest. 2017;152(3):502–9.
Nukui Y, Miyazaki Y, Masuo M, Okamoto T, Furusawa H, Tateishi T, Kishino M, Tateishi U, Ono J, Ohta S, Izuhara K. Periostin as a predictor of prognosis in chronic bird-related hypersensitivity pneumonitis. Allergology International. 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Anoop Nambiar reports research grants and personal fees from Boehringer-Ingelheim, Genentech/Roche, Veractye, Nitto Denko, and Galapagos outside of the submitted work.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Interstitial Lung Disease
Rights and permissions
About this article

Cite this article
Tam, W.S., Islam, T. & Nambiar, A.M. Hypersensitivity Pneumonitis (Including Environmental Assessment): Diagnosis and Management. Curr Pulmonol Rep 8, 131–138 (2019). https://doi.org/10.1007/s13665-019-00239-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13665-019-00239-6