
Abstract
Purpose of Review
Small-molecule kinase inhibitors (SMKIs) have revolutionized the management of malignant and autoimmune disorders. Emerging clinical reports point towards an increased risk for invasive fungal infections (IFIs) in patients treated with certain SMKIs. In this mini-review, we highlight representative examples of SMKIs that have been associated with or are expected to give rise to IFIs.
Recent Findings
The clinical use of the Bruton’s tyrosine kinase inhibitor ibrutinib as well as other FDA-approved SMKIs has been associated with IFIs. The fungal infection susceptibility associated with the clinical use of certain SMKIs underscores their detrimental effects on innate and adaptive antifungal immune responses.
Summary
The unprecedented development and clinical use of SMKIs is expected to give rise to an expansion of iatrogenic immunosuppressive factors predisposing to IFIs (and other opportunistic infections). Beyond increased clinical surveillance, better understanding of the pathogenesis of SMKI-associated immune dysregulation should help in devising improved risk stratification and prophylaxis strategies in vulnerable patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With ~5 million species, fungi constitute a large and diverse eukaryotic lineage consisting of species that range from the ecologically important saprotrophs that facilitate nutrient cycling to fungi responsible for large-scale loss of amphibians and bats (i.e., Batrachochytrium dendrobatidis, Geomyces destructans) [1, 2]. Humans have evolved to resist infections by most fungi; however, a small fraction of fungal pathogens such as the dermatophytes, the commensal yeast Candida, and environmental fungi such as the inhaled molds (primarily Aspergillus), and Cryptococcus species are common causes of infections in humans [3]. Infections of skin and nails by dermatophytes and of oral and genital mucosal surfaces by Candida species are the most common human fungal diseases [4], accounting for an estimated ~8.6 million outpatient visits, with an associated cost of ~$460 million per year in the USA alone [5, 6•]. Of greater clinical concern, life-threatening invasive infections by Candida, Aspergillus, Cryptococcus, and Pneumocystis carry mortality rates that exceed 50% despite administration of antifungal therapy, leading to > 1 million deaths worldwide per year [4].
Fungi did not emerge as major human pathogens until the late twentieth century [7], concurrently with the HIV/AIDS epidemic and major advances in modern medicine that has led to a significant expansion of patient populations with iatrogenic immunodeficiency [8]. With the introduction of broad-spectrum antibiotics for bacterial infections, myeloablative chemotherapy for malignancies, glucocorticoids and other immunomodulatory regimens for autoimmune diseases, and the progress in solid-organ transplantation (SOT) and hematopoietic stem cell transplantation (HSCT), modern medicine succeeded to change the natural history of many previously incurable diseases and extend the lives of millions of patients, yet at the price of compromising innate and/or adaptive immune functions [3].
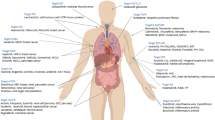
More recently, the advent of precision medicine therapies with novel small molecules targeting a variety of signaling kinases has revolutionized the treatment of malignancies and inflammatory diseases [9••]. However, many of these signaling kinase inhibitors also target key signaling pathways involved in host protection against fungi (and other pathogens) [9••]. Indeed, clinical reports describing an increased incidence of fungal infections associated with the clinical use of small-molecule kinase inhibitors (SMKIs) are now emerging [9••, 10••, 11••, 12••]. In light of the unprecedented rate of development of these compounds and their use in patients already at-risk for development of opportunistic infections, it is anticipated that we will witness an expansion of iatrogenic risk factors associated with invasive fungal (and other opportunistic) infections and new populations of patients with iatrogenic immunodeficiency in the coming years. In this mini-review, we highlight a few characteristic examples of SMKIs that have been associated with or are expected to give rise to fungal disease. A detailed review of all SMKIs leading to fungal disease and of non-fungal opportunistic infections that arise with SMKI treatment is beyond the scope of this report. Table 1 outlines the spectrum of reported SMKI-associated invasive fungal infections (IFIs) in humans.
SMKIs as Novel Iatrogenic Risk Factors for IFIs
Fungi belonging to five genera (Candida, Aspergillus, Cryptococcus, Pneumocystis, Mucorales) are responsible for > 90% of IFIs in humans [4, 53]. The host risk factors associated with these infections vary greatly depending on the fungal species. As such, the presence of implanted medical devices, the use of central venous catheters, neutropenia, broad-spectrum antibiotic use, and intra-abdominal surgery are known predisposing factors for invasive candidiasis, underscoring the importance of myeloid phagocytes and intact mucosal barrier function in preventing this infection [54]. In addition, patients with neutropenia and/or corticosteroid use in the setting of hematological malignancies, SOT, or HSCT, and patients with neutrophil dysfunction-associated primary immunodeficiencies such as chronic granulomatous disease are at risk for aspergillosis and mucormycosis [53, 55,56,57]. In contrast to the critical role of neutrophils for the control of the aforementioned infections, quantitative and/or qualitative defects in CD4+ T cells, such as with HIV infection, significantly enhance the risk for infections by Cryptococcus and Pneumocystis jirovecii [58,59,60]. Knowledge of these fungus-specific immune requirements for host defense is critical in understanding the pathogenesis and phenotypic expression of SMKI-associated IFIs in vulnerable patients.
Ibrutinib
An increasing number of clinical reports of IFIs have recently emerged with the use of inhibitors which target aberrantly active signaling pathways in patients with hematological malignancies or autoimmune diseases. A prominent example among these inhibitors is ibrutinib, a game-changing drug in the treatment of chronic lymphocytic leukemia (CLL). Beyond CLL, ibrutinib has also become a significant treatment modality for other B cell-targeted hematologic malignancies including mantle cell lymphoma, Waldenström macroglobulinemia, diffuse large B cell lymphoma, and primary central nervous system (CNS) lymphoma (PCNSL), as well as in HSCT recipients with graft-versus-host disease [9••, 61].
Ibrutinib is a covalent inhibitor of the Bruton’s tyrosine kinase (BTK), which is critical for B cell receptor signaling and promotes B cell development and survival. In lymphomas, targeting BTK via ibrutinib leads to inhibition of pro-survival signals and drives elimination of malignant cells [62]. Its use, however, has been associated with an increased incidence of invasive infections by a broad range of opportunistic fungi such Aspergillus, Fusarium, Mucorales, Cryptococcus, and Pneumocystis [9••]. Strikingly, in an ibrutinib-based trial of patients with primary CNS lymphoma (PCNSL), 39% of treated patients developed invasive aspergillosis and 11% developed Pneumocystis jirovecii pneumonia (PCP) [12••]. In another clinical trial involving CLL patients, ibrutinib use led to PCP in 5% of them despite adequate CD4 counts (> 500/ul), with an estimated incidence of 2.05 cases per 100 patient years [63•]. Additional retrospective analyses at four different clinical centers have also shown occurrence of IFIs associated with ibrutinib use (Table 1) [10••, 11••, 13•, 15•]. In contrast, in other settings of ibrutinib use, there has not been a significant association with IFIs. In view of the lack of prospective epidemiological studies in large cohorts of different patient groups, the absolute risk of IFI associated with ibrutinib is difficult to quantify. Collectively, the available clinical data suggest that BTK may play a critical role in protection against various fungi, which becomes essential in the setting of additional (host or iatrogenic) immunocompromising factors; however, mechanistic details of BTK-dependent antifungal immunity remain less clear.
BTK is expressed on all the hematopoietic cell types except for T cells and plasma cells [64]. In the case of Pneumocystis jirovecii, as B cells have been shown to play an important role in protection during PCP via priming of anti-Pneumocystis T cell responses [65,66,67], a direct role of ibrutinib on inhibiting B cell-targeted BTK is plausible for the development of PCP. Moreover, considering the central role of CD4+ T cells in protection against PCP, an off-target inhibition of T cell-targeted kinases such as ITK (interleukin-2-inducible T cell kinase) by ibrutinib is also possible in driving PCP susceptibility. Additionally, it is likely that BTK exerts functions on myeloid antigen-presenting cells such as dendritic cells (DCs) and macrophages for protective anti-Pneumocystis immunity, given the crucial role that DCs play in CD4+ T cell priming and that macrophages play in efficient intracellular Pneumocystis clearance [68]. Therefore, further investigation is required to fully understand the immunopathogenesis behind PCP infection susceptibility in the context of BTK pharmacological inhibition.
Similar to PCP, CD4+ T cells are also of central importance for orchestrating protection against cryptococcosis, as evident by the emergence of cryptococcal infections during the HIV/AIDS epidemic [69]. Cryptococcosis is also prevalent in patients in other acquired immunodeficiencies that affect T cell function such as in patients receiving SOT or HSCT, in hematological malignancies, or in patients on systemic corticosteroid therapy [70]. Studies using mouse models of subacute or chronic cryptococcal infections have also revealed the important roles of monocyte-derived DCs in CD4+ T cell priming, their skewing towards a Th1-phenotype, and consequently the roles of Th1 cytokines in macrophage activation towards the M1 phenotype for effective fungal clearance [71,72,73]. Consistently, patients with mutations in IL12 or the IL12 receptor, in CD40L, or those carrying neutralizing autoantibodies against IFN-γ and granulocyte-macrophage colony-stimulating factor (GM-CSF) have also been reported to develop cryptococcosis [74], highlighting a critical role for the cross-talk between T cells and macrophages in sterilizing cryptococcal immunity. It is therefore possible that ibrutinib-associated development of cryptococcosis may arise from either off-target effects on BTK-related kinases expressed on T cells and/or a direct effect of ibrutinib on BTK signaling on myeloid phagocytes. Indeed, reduced phagocytosis by alveolar macrophages, decreased levels of anti-Cryptococcus IgM, and increased susceptibility to Cryptococcus infection has also been reported in studies using X-linked immunodeficient mice carrying a mutation in BTK [75]. Ultimately, it is apparent that host defense against Cryptococcus requires the intricate and synchronized interplay of both cell-mediated and humoral immunity, with both myeloid phagocytes as well as T cells and even B cells critical for clearance of the fungus [76]. In view of BTK’s almost universal expression among immune cells and its major role in regulating development and multiple effector functions, direct inhibition with ibrutinib could indeed affect susceptibility to infection and more studies are needed to elucidate the cell type-specific effects on ibrutinib in inhibiting anticryptococcal host defense.
Unlike Pneumocystis and Cryptococcus, anti-Aspergillus host defense primarily relies on myeloid phagocytes [56, 77]. Instead, cells of the lymphoid lineage are dispensable for host protection as mice or patients lacking lymphoid cells are not susceptible to invasive aspergillosis [56, 78••]. Owing to the expression of BTK on myeloid cells, inhibition of BTK-dependent effector functions of myeloid cells by ibrutinib may compromise anti-Aspergillus defense. Indeed, we have demonstrated that myeloid phagocyte-specific conditional BTK knockout mice are susceptible to invasive pulmonary aspergillosis and phenocopy the susceptibility to the infection observed in global BTK-deficient mice (Desai and Zarakas et al., in preparation) [12••]. Furthermore, it has been shown that, in murine and human monocyte-derived macrophages, BTK functions downstream of Dectin-1 and TLR9 fungal sensing to promote NFAT/NFĸB-dependent TNF production in the setting of ex vivo challenge with A. fumigatus [79••, 80•]. NFAT signaling in myeloid phagocytes also regulates pentraxin production and anti-Aspergillus host defense [81]. However, the precise myeloid cellular subsets and the molecular mechanisms responsible for BTK-dependent Aspergillus clearance in vivo remain unclear and are a subject of ongoing research investigation.
These collective data indicate a crucial role for BTK in antifungal defense; however, patients with X-linked agammaglobulinemia (XLA), who harbor mutations in BTK rarely develop fungal infections. In fact, only two cases of fungal infections have been reported in XLA patients thus far; one with PCP and one with invasive aspergillosis [82, 83]. These observations indicate that a constellation of predisposing factors, in addition to the acute pharmacological inhibition by ibrutinib per se, may impact the incidence of IFIs in ibrutinib-treated patients. Such factors include, but are not limited to, the underlying lymphoid malignancy and its status (active versus in remission), additional genetic predisposition via polymorphisms in immune-related genes, pharmacogenetic variation that may result in greater ibrutinib exposures, co-administration of other pharmacological agents and their impacts on the immune status, the age of the patient [84•], and the extent of fungal exposure including the inoculum and fungal strain. In addition, compensatory mechanisms may be operational in XLA patients to overcome long-term, early-onset BTK-dependent inhibition of antifungal immune effector mechanisms, as opposed to the acute pharmacological BTK inhibition conferred by ibrutinib. With the advent of second-generation BTK inhibitors that are expected to have greater specificity for BTK over other non-BTK kinases, it will be important to carefully define the incidence of IFIs relative to that of ibrutinib.
Ruxolitinib
Ruxolitinib, an inhibitor of Janus-associated kinases (JAK) 1 and 2, was initially approved by the FDA in 2011 for the treatment of myelofibrosis and was later approved for polycythemia vera in 2014 [85]. The pathogenesis of myelofibrosis involves dysregulation of signaling through JAK1/2-signal transducer and activator of transcription (STAT) pathways, leading to reactive bone marrow fibrosis, splenomegaly, extramedullary hematopoiesis, and increased risk for leukemia progression and decreased survival [86,87,88]. Ruxolitinib-mediated JAK1/2 inhibition has shown marked and durable clinical benefits in terms of reductions in splenomegaly and disease-related symptoms [89,90,91,92].
In the initial randomized clinical trials, ruxolitinib treatment exerted hematological side effects, mainly dose-related anemia, thrombocytopenia, and neutropenia [90, 93], while data on infections were not initially systematically captured [89, 90, 94,95,96], with the exception of a signal for herpes zoster virus infections [94]. Since ruxolitinib came into the market, multiple case reports have surfaced detailing infectious complications caused by viruses and bacteria [34, 96,97,98,99,100,101,102,103]. As outlined in Table 1, reports of opportunistic fungal infections have also emerged with ruxolitinib (and other JAK/STAT inhibitor) use.
Given the prominent role of JAK/STAT signaling downstream of diverse cytokine receptors, increasing evidence suggests that ruxolitinib-dependent JAK1/2 inhibition exerts immunosuppressive effects [104], leading to enhanced susceptibility to infection. In the case of fungal infections, the importance of JAK-STAT signaling downstream of type I–III interferons and other cytokines in host immune defense is beginning to unravel. For example, in neutrophils, cell-intrinsic STAT1 activation via IFN-λ/IFNLR1 signaling leads to reactive oxygen species production for efficient Aspergillus clearance [78••]. Additionally, C. neoformans-dependent transcriptional activation of JAK/STAT signaling in monocytes has been reported [105]. Furthermore, given the central role of JAK-STAT signaling in T cell and macrophage physiology [106] and effector functions [107•], direct ruxolitinib-derived functional impairment of the T cell-macrophage cross-talk leading to cryptococcosis and PCP is likely. More research is required to elucidate the detrimental antifungal immune effects conferred by ruxolitinib and other JAK-STAT inhibitors leading to opportunistic (including fungal) infections. In addition, expanded use of JAK/STAT inhibitors in patients with additional immunosuppressive factors (e.g., transplant recipients) could result in higher number of IFIs.
Sorafenib
Sorafenib is an oral multi-kinase inhibitor of cell surface tyrosine kinase receptors and intracellular serine/threonine kinases in the RAS/mitogen-activated protein kinase (MAPK) cascade. Sorafenib was approved by the FDA for the treatment of advanced renal cell carcinoma in 2005, unresectable hepatocellular carcinoma in 2007, and metastatic differentiated thyroid cancer in 2013 [108,109,110]. By blocking the activity of Raf-1, BRAF and kinases in the RAS/ extracellular signal-regulated kinase (ERK), and mitogen-activated kinase/ERK (RAS/RAF/MEK/ERK) signaling pathway, sorafenib inhibits tumor proliferation and survival and induces tumor cell apoptosis [109,110,111]. In addition, sorafenib inhibits angiogenesis through vascular endothelial growth factor receptor (VEGFR) 1, 2, and 3; platelet-derived growth factor receptor β (PDGFR-β); and other tyrosine kinases [109,110,111]. Currently, multiple ongoing clinical trials are examining the therapeutic potential of sorafenib for a plethora of cancers (clinicaltrials.gov). Potential beneficial outcomes of sorafenib in the treatment of acute myelogenous leukemia [112, 113] and salivary tumors have also been reported [38].
Adverse events in sorafenib-treated patients are predominantly gastrointestinal, constitutional, or dermatologic in nature, including diarrhea, weight loss, and hand–foot skin reactions [109]. More recently, cases of sorafenib-induced acute interstitial pneumonia [114] and other cutaneous side effects were reported [115] [116]. As summarized in Table 1, IFIs have been associated with the use of sorafenib, including three cases with invasive aspergillosis [28, 38,39,40]. Notably, two cases appeared in the absence of concurrent immunosuppressive treatment with chemotherapy or corticosteroids within the last month prior to the fungal infection diagnosis, implicating sorafenib alone for the increased susceptibility of infection [28, 38]. Furthermore, sorafenib-treated patients were reported to develop mucocutaneous fungal infections caused by Candida and Rhodotorula mucilaginosa yeasts [117, 118]. In the context of acute myeloid leukemia treated with sorafenib, fungal lung nodules and fungal pneumonia have also been reported [112, 113].
Multiple immune modulating functions of sorafenib can potentially account for the increased risk for mucosal fungal disease and IFIs. The RAS associated with diabetes (RAD)/MAPK/ERK signaling pathway, a major target of sorafenib, is important for antifungal effector functions in phagocytes against fungal species [119, 120]. In addition, ERK signaling regulates killing of Aspergillus by macrophages independently of TLR signaling [119]. Moreover, by interfering with phosphoinositide 3-kinase (PI3), MAP kinases and NF-kB signaling, sorafenib inhibits DC function by inducing apoptosis, and by impairing antigen presentation, it results in decreased T cell responses, which may underlie the mucosal fungal susceptibility [121]. In addition, RAF-dependent activation of JAK/STAT signaling may also be inhibited by sorafenib [122], with potential negative effects on antifungal immunity, as mentioned above.
Fostamatinib
Fostamatinib is an oral spleen tyrosine kinase (Syk) inhibitor developed for the treatment of immune thrombocytopenic purpura (ITP), autoimmune hemolytic anemia (AHA), and IgA nephropathy. It was recently approved by the FDA for the treatment of adult treatment-refractory ITP in 2018, as it was shown to inhibit platelet destruction and achieve durable clinical responses [123, 124]. Syk is a principal regulatory kinase that acts downstream of multiple fungal-sensing pattern recognition receptors of the C-type lectin receptor family [77]. Upon activation, Syk-dependent signaling engages the adaptor caspase recruitment domain family member 9 (CARD9), which assembles with B cell CLL/lymphoma 10 (BCL10) and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) to relay downstream antifungal responses, ultimately leading to the induction of ERK and NF-kB and the production of inflammatory mediators such as IL-6, IL-12, GM-CSF, TNFa, and IL-1β [77].
Patients with inherited CARD9 deficiency develop specific and severe susceptibility to fungal infections [125•]. Specifically, CARD9-deficient patients suffer from spontaneous fungal infections, predominantly localized to the oral mucosa, central nervous system (CNS), bone, and subcutaneous tissues, caused by Candida, Aspergillus, Exophiala, Phialophora, and other phaeohyphomycetes [125•]. The mechanisms of Syk-CARD9-dependent antifungal immunity are now being elucidated. In the CNS, CARD9-dependent signaling is necessary for protective neutrophil recruitment via mechanisms that relate to induction of protective factors within the CNS, not neutrophil-intrinsic survival or chemotaxis [126••]. We recently showed that the secreted fungal secreted toxin candidalysin acts on brain-resident microglia, in a CARD9-dependent manner to induce transcriptional activation and inflammasome-dependent production of IL-1β, which in turn drives microglial CXCL1 production to recruit protective CXCR2+ neutrophils in the Candida-infected brain [127••]. Indeed, CARD9-deficient patients have absent CXCL1 in the Candida-infected cerebrospinal fluid and do not mobilize neutrophils in the fungal-infected CNS [126••]. Additional detrimental effects of CARD9 deficiency on neutrophil effector function include a selective defect in killing of unopsonized Candida yeast forms, which may also contribute to the patient fungal susceptibility, by compromising the function of the few neutrophils that traffic into the infected CNS [128].
CARD9-deficient patients were also reported to develop extrapulmonary aspergillosis, associated with a defect in neutrophil accumulation in the infected tissue [129•]. CARD9-dependent induction of IL-17 may underlie the susceptibility to mucocutaneous fungal disease [130]. Thus, owing to the central role of Syk-CARD9 signaling in antifungal host defense, careful surveillance of fostamatinib-treated patients for the development of fungal disease is warranted. So far, a case of vaginal yeast infection in a fostamatinib-treated woman was described [41]. There are currently 44 clinical trials of fostamatinib treatment registered in clinicaltrials.gov, including in the management of conditions that already predispose patients to fungal disease (e.g., leukemia, graft-versus-host disease). Besides the potential direct effects of fostamatinib in compromising antifungal immune responses, neutropenia can occur in a small proportion of fostamatinib-treated patients, further increasing the risk for fungal infections [124]. Data from the ongoing clinical trials and post-market surveillance will shed light on the degree by which fostamatinib treatment in humans may pose a risk of increased susceptibility to IFIs, as it would be predicted based on the inherited CARD9 deficiency.
Conclusions
A recent surge in the development and clinical use of SMKIs has undoubtedly changed the treatment paradigm of serious, often fatal, human diseases. Some of these molecules can also target critical immune surveillance pathways, creating a permissive environment for fungi (and other opportunistic pathogens) to cause disease. With the expanding indications and the unprecedented rate of development of these compounds, new populations of patients with predicted or unpredicted iatrogenic immunosuppression may develop, requiring increased clinical surveillance for opportunistic infections and timely reporting. Real-time epidemiological data and case-control studies to identify the true risk of individual SMKIs for IFIs in the real world, out of the setting of selected patients participating in phase II/III trials, are missing and are urgently needed. More research into the physiological antifungal effector signaling pathways and the detrimental effects that SMKIs have on the innate and adaptive immune system should allow for better risk stratification and prophylaxis of susceptible patients. Finally, there is an unmet need for development of functional immune assays that will allow for an estimate of the net state of immunodeficiency of the individual patient who is a candidate for or is receiving SMKI therapy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Skerratt LF, Berger L, Speare R, Cashins S, McDonald KR, Phillott AD, et al. Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. Ecohealth. 2007;4(2):125–34. https://doi.org/10.1007/s10393-007-0093-5.
Frick WF, Pollock JF, Hicks AC, Langwig KE, Reynolds DS, Turner GG, et al. An emerging disease causes regional population collapse of a common North American bat species. Science (New York, NY). 2010;329(5992):679–82. https://doi.org/10.1126/science.1188594.
Kohler JR, Hube B, Puccia R, Casadevall A, Perfect JR. Fungi that infect humans. Microbiol Spectrum. 2017;5(3). https://doi.org/10.1128/microbiolspec.FUNK-0014-2016.
Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. Hidden killers: human fungal infections. Sci Transl Med. 2012;4(165):165rv13. https://doi.org/10.1126/scitranslmed.3004404.
Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51(Suppl 4):2–15. https://doi.org/10.1111/j.1439-0507.2008.01606.x.
• Benedict K, Jackson BR, Chiller T, Beer KD. Estimation of direct healthcare costs of fungal diseases in the United States. Clin Infect Dis. 2018. https://doi.org/10.1093/cid/ciy776 Recent study using data from large insurance claims databases that sheds light on the profound burden that fungal diseases impose on the healthcare system of the USA.
Robert VA, Casadevall A. Vertebrate endothermy restricts most fungi as potential pathogens. J Infect Dis. 2009;200(10):1623–6. https://doi.org/10.1086/644642.
Lionakis MS, Levitz SM. Host control of fungal infections: lessons from basic studies and human cohorts. Annu Rev Immunol. 2018;36:157–91. https://doi.org/10.1146/annurev-immunol-042617-053318.
•• Chamilos G, Lionakis MS, Kontoyiannis DP. Call for action: invasive fungal infections associated with Ibrutinib and other small molecule kinase inhibitors targeting immune signaling pathways. Clin Infect Dis. 2018;66(1):140–8. https://doi.org/10.1093/cid/cix687 A comprehensive review of SMKIs outlining their association with IFIs .
•• Ghez D, Calleja A, Protin C, Baron M, Ledoux MP, Damaj G, et al. Early-onset invasive aspergillosis and other fungal infections in patients treated with ibrutinib. Blood. 2018;131(17):1955–9. https://doi.org/10.1182/blood-2017-11-818286 A retrospective study of patients treated with ibrutinib during the period 2013–2017, demostrating increased incidence of IFIs, particularly IA with frequent cerebral involvement.
•• Varughese T, Taur Y, Cohen N, Palomba ML, Seo SK, Hohl TM, et al. Serious infections in patients receiving ibrutinib for treatment of lymphoid cancer. Clin Infect Dis. 2018;67(5):687–92. https://doi.org/10.1093/cid/ciy175 A retrospective analysis of lymphoid cancer patients receiving ibrutinib during a 5-year period, indicating ibrutinib as a major risk factor for serious infections, including IFIs.
•• Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, et al. Inhibition of B cell receptor signaling by ibrutinib in primary CNS lymphoma. Cancer Cell. 2017;31(6):833–43.e5. https://doi.org/10.1093/cid/ciy175 Important paper showing promising results of a Phase Ib study of ibrutinib use for the treatment of PCNSL. The authors also highlighted the remarkable 39% incidence of Aspergillus infections associated with ibrutinib therapy .
• Rogers KA, Mousa L, Zhao Q, Bhat SA, Byrd JC, El Boghdadly Z, et al. Incidence of opportunistic infections during ibrutinib treatment for B-cell malignancies. Leukemia. 2019. https://doi.org/10.1038/s41375-019-0481-1 A single institution retrospective study of opportunistic infections (OIs) in ibrutinib-treated patients, in which IFIs accounted for the majority of opportunistic infections.
Rogers KA, Luay M, Zhao QH, Wiczer T, Levine L, Zeinab E, et al. Incidence and type of opportunistic infections during Ibrutinib treatment at a single academic center. Blood. 2017;130.
• Barbosa CC, DeAngelis LM, Grommes C. Ibrutinib associated infections: a retrospective study. 2017;35(15_suppl):e19020-e. https://doi.org/10.1200/JCO.2017.35.15_suppl.e19020 A retrospective analysis of patients treated with ibrutinib for various lymphomas between 4/2014–11/2016. The authors reported 7 cases of IFIs among 200 treated patients.
Sun K, Kasparian S, Iyer S, Pingali SR. Cryptococcal meningoencephalitis in patients with mantle cell lymphoma on ibrutinib. Ecancermedicalscience. 2018;12:836.
Swan CD, Gottlieb T. Cryptococcus neoformans empyema in a patient receiving ibrutinib for diffuse large B-cell lymphoma and a review of the literature. BMJ Case Rep. 2018;2018. https://doi.org/10.3332/ecancer.2018.836.
Grossi O, Pineau S, Sadot-Lebouvier S, Hay B, Delaunay J, Miailhe AF, et al. Disseminated mucormycosis due to Lichtheimia corymbifera during ibrutinib treatment for relapsed chronic lymphocytic leukaemia: a case report. Clin Microbiol Infect. 2019;25(2):261–3. https://doi.org/10.1016/j.cmi.2018.10.004.
Nasir T, Lee C, Lawrence AS, Brown JS. Invasive aspergillosis complicating treatment with tyrosine kinase inhibitors. BMJ Case Rep. 2019;12(1):e226121. https://doi.org/10.1136/bcr-2018-226121.
Faisal MS, Shaikh H, Khattab A, Albrethsen M, Fazal S. Cerebral aspergillosis in a patient on ibrutinib therapy-a predisposition not to overlook. J Oncol Pharm Pract. 2018:1078155218788717. https://doi.org/10.1177/1078155218788717.
Beresford R, Dolot V, Foo H. Cranial aspergillosis in a patient receiving ibrutinib for chronic lymphocytic leukemia. Med Mycol Case Rep. 2019;24:27–9. https://doi.org/10.1016/j.mmcr.2019.02.005.
Stephens DM, Byrd JC. How I manage ibrutinib intolerance and complications in patients with chronic lymphocytic leukemia. Blood. 2019;133(12):1298–307. https://doi.org/10.1182/blood-2018-11-846808.
McCarter SJ, Vijayvargiya P, Sidana S, Nault AM, Lane CE, Lehman JS, et al. A case of ibrutinib-associated aspergillosis presenting with central nervous system, myocardial, pulmonary, intramuscular, and subcutaneous abscesses. Leuk Lymphoma. 2019;60(2):559–61. https://doi.org/10.1080/10428194.2018.1494271.
Pouvaret A, Guery R, Montillet M, Molina TJ, Dureault A, Bougnoux ME, et al. Concurrent cerebral aspergillosis and abdominal mucormycosis during ibrutinib therapy for chronic lymphocytic leukaemia. Clin Microbiol Infect. 2019;25:771–3. https://doi.org/10.1016/j.cmi.2019.01.016.
Wilson PA, Melville K. Disseminated cryptococcal infection in a patient receiving acalabrutinib for chronic lymphocytic leukemia. 9000;Publish Ahead of Print. https://doi.org/10.1097/IPC.0000000000000722, 2019.
Wysham NG, Sullivan DR, Allada G. An opportunistic infection associated with ruxolitinib, a novel janus kinase 1,2 inhibitor. Chest. 2013;143(5):1478–9. https://doi.org/10.1378/chest.12-1604.
Lee SC, Feenstra J, Georghiou PR. Pneumocystis jiroveci pneumonitis complicating ruxolitinib therapy. BMJ Case Rep. 2014;2014:bcr2014204950. https://doi.org/10.1136/bcr-2014-204950.
Chan JF, Chan TS, Gill H, Lam FY, Trendell-Smith NJ, Sridhar S, et al. Disseminated infections with Talaromyces marneffei in non-AIDS patients given monoclonal antibodies against CD20 and kinase inhibitors. Emerg Infect Dis. 2015;21(7):1101–6. https://doi.org/10.3201/eid2107.150138.
Chen CC, Chen YY, Huang CE. Cryptococcal meningoencephalitis associated with the long-term use of ruxolitinib. Ann Hematol. 2016;95(2):361–2. https://doi.org/10.1007/s00277-015-2532-7.
Hirano A, Yamasaki M, Saito N, Iwato K, Daido W, Funaishi K, et al. Pulmonary cryptococcosis in a ruxolitinib-treated patient with primary myelofibrosis. Respir Med Case Rep. 2017;22:87–90. https://doi.org/10.1016/j.rmcr.2017.06.015.
Polverelli N, Breccia M, Benevolo G, Martino B, Tieghi A, Latagliata R, et al. Risk factors for infections in myelofibrosis: role of disease status and treatment. A multicenter study of 507 patients. Am J Hematol. 2017;92(1):37–41. https://doi.org/10.1002/ajh.24572.
Liu J, Mouhayar E, Tarrand JJ, Kontoyiannis DP. Fulminant Cryptococcus neoformans infection with fatal pericardial tamponade in a patient with chronic myelomonocytic leukaemia who was treated with ruxolitinib: case report and review of fungal pericarditis. Mycoses. 2018;61(4):245–55. https://doi.org/10.1111/myc.12735.
Moruno-Rodriguez A, Sanchez-Vicente JL, Rueda-Rueda T, Lechon-Caballero B, Munoz-Morales A, Lopez-Herrero F. Invasive aspergillosis manifesting as retinal necrosis in a patient treated with ruxolitinib. Arch Soc Esp Oftalmol. 2019;94(5):237–41. https://doi.org/10.1016/j.oftal.2018.12.006.
Dioverti MV, Abu Saleh OM, Tande AJ. Infectious complications in patients on treatment with ruxolitinib: case report and review of the literature. Infect Dis (London, Engl). 2018;50(5):381–7. https://doi.org/10.1080/23744235.2017.1390248.
Prakash K, Richman D. A case report of disseminated histoplasmosis and concurrent cryptococcal meningitis in a patient treated with ruxolitinib. BMC Infect Dis. 2019;19(1):287. https://doi.org/10.1186/s12879-019-3922-6.
Chakrabarti A, Sood N. Cryptococcal meningitis in an immunocompetent patient with primary myelofibrosis on long-term ruxolitinib: report of a rare case and review of literature. memo – Mag Eur Med Oncol. 2018;11(4):348–50. https://doi.org/10.1007/s12254-018-0435-8.
• Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. 2018;128(7):3041–52. https://doi.org/10.1172/JCI98814 This study reported fungal infections in 4 out of 18 baricitinib-treated patients with interferonopathies .
Locati LD, Perrone F, Cortelazzi B, Bergamini C, Bossi P, Civelli E, et al. A phase II study of sorafenib in recurrent and/or metastatic salivary gland carcinomas: translational analyses and clinical impact. Eur J Cancer (Oxford, Engl: 1990). 2016;69:158–65. https://doi.org/10.1016/j.ejca.2016.09.022.
Bazaz R, Denning DW. Subacute invasive aspergillosis associated with sorafenib therapy for hepatocellular carcinoma. Clin Infect Dis. 2018;67(1):156–7. https://doi.org/10.1093/cid/ciy038.
Kloos RT, Ringel MD, Knopp MV, Hall NC, King M, Stevens R, et al. Phase II trial of sorafenib in metastatic thyroid cancer. J Clin Oncol. 2009;27(10):1675–84. https://doi.org/10.1200/JCO.2008.18.2717.
Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–85. https://doi.org/10.1182/blood-2009-08-236471.
Al-Ameri A, Kantarjian H, Borthakur G, Bahceci E, Szatrowski T, Damokosh A, et al. Opportunistic infections are uncommon with dasatinib in patients with chronic myeloid leukemia in chronic phase (CML-CP). Blood. 2009;114(22):1120.
Chang H, Hung YS, Chou WC. Pneumocystis jiroveci pneumonia in patients receiving dasatinib treatment. Int J Infect Dis. 2014;25:165–7. https://doi.org/10.1016/j.ijid.2014.04.030.
Su CX, Ren SX, Li XF, Hou LK, Zhou CC. Pseudo-progression in a patient with lung adenocarcinoma and ALK fusion who responded to crizotinib. Int J Clin Exp Med. 2016;9(6):12290–3.
Deiana L, Grisanti S, Ferrari V, Tironi A, Brugnoli G, Ferrari L, et al. Aspergillosis superinfection as a cause of death of crizotinib-induced interstitial lung disease successfully treated with high-dose corticosteroid therapy. Case Rep Oncol. 2015;8(1):169–73. https://doi.org/10.1159/000381209.
Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18(8):1061–75. https://doi.org/10.1016/S1470-2045(17)30416-3.
Crisan AM, Ghiaur A, Stancioaca MC, Bardas A, Ghita C, Manea CM, et al. Mucormycosis during imatinib treatment: case report. J Med Life. 2015;8(3):365–70.
Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110delta, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390–7. https://doi.org/10.1182/blood-2013-11-535047.
Kim DY, Joo YD, Lim SN, Kim SD, Lee JH, Lee JH, et al. Nilotinib combined with multiagent chemotherapy for newly diagnosed Philadelphia-positive acute lymphoblastic leukemia. Blood. 2015;126(6):746–56. https://doi.org/10.1182/blood-2015-03-636548.
Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre PD, Paquette R, Chuah C, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. 2018;132(4):393–404. https://doi.org/10.1182/blood-2016-09-739086.
Visvardis EE, Gao F, Paes MN, Duprez O, Waxman J. Lung aspergillosis in renal cell carcinoma patient treated with sunitinib. QJM. 2012;105(7):689–92. https://doi.org/10.1093/qjmed/hcr091.
Kim YW, Lee HW, Cho J, Choi HS, Lee J, Park SS, et al. Conversion of aspergilloma to chronic necrotizing pulmonary aspergillosis following treatment with sunitinib: a case report. Oncol Lett. 2016;12(5):3472–4. https://doi.org/10.3892/ol.2016.5052.
Bongomin F, Gago S, Oladele RO, Denning DW. Global and multi-national prevalence of fungal diseases-estimate precision. J Fungi (Basel). 2017;3(4). https://doi.org/10.3390/jof3040057.
Pappas PG, Lionakis MS, Arendrup MC, Ostrosky-Zeichner L, Kullberg BJ. Invasive candidiasis. Nat Rev Dis Primers. 2018;4:18026. https://doi.org/10.1038/nrdp.2018.26.
Pagano L, Caira M, Candoni A, Offidani M, Fianchi L, Martino B, et al. The epidemiology of fungal infections in patients with hematologic malignancies: the SEIFEM-2004 study. Haematologica. 2006;91(8):1068–75.
Hohl TM. Immune responses to invasive aspergillosis: new understanding and therapeutic opportunities. Curr Opin Infect Dis. 2017;30(4):364–71. https://doi.org/10.1097/QCO.0000000000000381.
Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, et al. Common severe infections in chronic granulomatous disease. Clin Infect Dis. 2015;60(8):1176–83. https://doi.org/10.1093/cid/ciu1154.
Rajasingham R, Smith RM, Park BJ, Jarvis JN, Govender NP, Chiller TM, et al. Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect Dis. 2017;17(8):873–81. https://doi.org/10.1016/S1473-3099(17)30243-8.
Richardson M, Lass-Florl C. Changing epidemiology of systemic fungal infections. Clin Microbiol Infect. 2008;14(Suppl 4):5–24. https://doi.org/10.1111/j.1469-0691.2008.01978.x.
Maertens J, Vrebos M, Boogaerts M. Assessing risk factors for systemic fungal infections. Eur J Cancer Care. 2001;10(1):56–62. https://doi.org/10.1046/j.1365-2354.2001.00241.x.
Miklos D, Cutler CS, Arora M, Waller EK, Jagasia M, Pusic I, et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood. 2017;130(21):2243–50. https://doi.org/10.1182/blood-2017-07-793786.
Burger JA, Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. 2018;18(3):148–67. https://doi.org/10.1038/nrc.2017.121.
• Ahn IE, Jerussi T, Farooqui M, Tian X, Wiestner A, Gea-Banacloche J. Atypical Pneumocystis jirovecii pneumonia in previously untreated patients with CLL on single-agent ibrutinib. Blood. 2016;128(15):1940–3. https://doi.org/10.1182/blood-2016-06-722991 A study reporting incidence of PJP in 5% of patients on ibrutinib monotherapy for CLL.
Smith CI, Baskin B, Humire-Greiff P, Zhou JN, Olsson PG, Maniar HS, et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J Immunol (Baltimore, Md : 1950). 1994;152(2):557–65.
Kolls JK. An emerging role of B cell immunity in susceptibility to Pneumocystis pneumonia. Am J Respir Cell Mol Biol. 2017;56(3):279–80. https://doi.org/10.1165/rcmb.2016-0360ED.
Hoving JC, Kolls JK. New advances in understanding the host immune response to Pneumocystis. Curr Opin Microbiol. 2017;40:65–71. https://doi.org/10.1016/j.mib.2017.10.019.
Martin-Garrido I, Carmona EM, Specks U, Limper AH. Pneumocystis pneumonia in patients treated with rituximab. Chest. 2013;144(1):258–65. https://doi.org/10.1378/chest.12-0477.
Kelly MN, Shellito JE. Current understanding of Pneumocystis immunology. Future Microbiol. 2010;5(1):43–65. https://doi.org/10.2217/fmb.09.116.
Hajjeh RA, Brandt ME, Pinner RW. Emergence of cryptococcal disease: epidemiologic perspectives 100 years after its discovery. Epidemiol Rev. 1995;17(2):303–20. https://doi.org/10.1093/oxfordjournals.epirev.a036195.
Sloan DJ, Parris V. Cryptococcal meningitis: epidemiology and therapeutic options. Clin Epidemiol. 2014;6:169–82. https://doi.org/10.2147/CLEP.S38850.
Osterholzer JJ, Chen GH, Olszewski MA, Curtis JL, Huffnagle GB, Toews GB. Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6Chigh monocytes. J Immunol (Baltimore, Md : 1950). 2009;183(12):8044–53. https://doi.org/10.4049/jimmunol.0902823.
Osterholzer JJ, Chen GH, Olszewski MA, Zhang YM, Curtis JL, Huffnagle GB, et al. Chemokine receptor 2-mediated accumulation of fungicidal exudate macrophages in mice that clear cryptococcal lung infection. Am J Pathol. 2011;178(1):198–211. https://doi.org/10.1016/j.ajpath.2010.11.006.
Osterholzer JJ, Curtis JL, Polak T, Ames T, Chen GH, McDonald R, et al. CCR2 mediates conventional dendritic cell recruitment and the formation of bronchovascular mononuclear cell infiltrates in the lungs of mice infected with Cryptococcus neoformans. J Immunol (Baltimore, Md : 1950). 2008;181(1):610–20. https://doi.org/10.4049/jimmunol.181.1.610.
Lionakis MS, Netea MG, Holland SM. Mendelian genetics of human susceptibility to fungal infection. Cold Spring Harb Perspect Med. 2014;4(6). https://doi.org/10.1101/cshperspect.a019638.
Szymczak WA, Davis MJ, Lundy SK, Dufaud C, Olszewski M, Pirofski LA. X-linked immunodeficient mice exhibit enhanced susceptibility to Cryptococcus neoformans infection. mBio. 2013;4(4). https://doi.org/10.1128/mBio.00265-13.
Rohatgi S, Pirofski LA. Host immunity to Cryptococcus neoformans. Future Microbiol. 2015;10(4):565–81.
Lionakis MS, Iliev ID, Hohl TM. Immunity against fungi. JCI Insight. 2017;2(11):565–81. https://doi.org/10.2217/fmb.14.132.
•• Espinosa V, Dutta O, McElrath C, Du P, Chang YJ, Cicciarelli B, et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci Immunol. 2017;2(16). https://doi.org/10.1126/sciimmunol.aan5357. This study describes how neutrophil-intrinsic STAT1 activation via IFN-λ/IFNLR1 signaling leads to reactive oxygen species production for efficient Aspergillus clearance. Given the association of IFIs with ruxolitinib (JAK1/2 inhibitor) use, the findings from this study are highly relevant.
•• Bercusson A, Colley T, Shah A, Warris A, Armstrong-James D. Ibrutinib blocks Btk-dependent NF-kB and NFAT responses in human macrophages during Aspergillus fumigatus phagocytosis. Blood. 2018;132(18):1985–8. https://doi.org/10.1182/blood-2017-12-823393 This study demonstrates that ibrutinib-mediated blockade of BTK decreases A. fumigatus-induced NF-kB and NFAT activation in human macrophages.
• Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol Med. 2015;7(3):240–58. https://doi.org/10.15252/emmm.201404556 This study demonstrates that phagocytosis of A. fumigatus activates NFAT in BTK-dependent manner.
Zelante T, Wong AY, Mencarelli A, Foo S, Zolezzi F, Lee B, et al. Impaired calcineurin signaling in myeloid cells results in downregulation of pentraxin-3 and increased susceptibility to aspergillosis. Mucosal Immunol. 2017;10(2):470–80. https://doi.org/10.1038/mi.2016.52.
Kanegane H, Nakano T, Shimono Y, Zhao M, Miyawaki T. Pneumocystis jiroveci pneumonia as an atypical presentation of X-linked agammaglobulinemia. Int J Hematol. 2009;89(5):716–7. https://doi.org/10.1007/s12185-009-0322-5.
Nishi K, Kawai T, Kubota M, Ishiguro A, Onodera M. X-linked agammaglobulinemia complicated with pulmonary aspergillosis. Pediatr Int. 2018;60(1):90–2. https://doi.org/10.1111/ped.13453.
• Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, et al. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-Cell diffuse large B-Cell lymphoma. J Clin Oncol. 2019:Jco1802403. https://doi.org/10.1200/JCO.18.02403. A paper outlining the patient age as a possible determining factor for clinical outcomes in lymphoma patients treated with ibrutinib-based chemotherapy. Higher rates of infection and worse outcomes were demonstrated in patients 60 years or older. Interestingly, Aspergillus infections were only reported in this group of patients.
Raedler LA. Jakafi (Ruxolitinib): first FDA-approved medication for the treatment of patients with polycythemia vera. Am Health Drug Benefits. 2015;8(Spec Feature):75–9.
Tefferi A. Primary myelofibrosis: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2016;91(12):1262–71. https://doi.org/10.1002/ajh.24592.
Gregory SA, Mesa RA, Hoffman R, Shammo JM. Clinical and laboratory features of myelofibrosis and limitations of current therapies. Clin Adv Hematol Oncol. 2011;9(9 Suppl 22):1–16.
Mughal TI, Vaddi K, Sarlis NJ, Verstovsek S. Myelofibrosis-associated complications: pathogenesis, clinical manifestations, and effects on outcomes. Int J Gen Med. 2014;7:89–101.
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. https://doi.org/10.1056/NEJMoa1110557.
Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–98. https://doi.org/10.1056/NEJMoa1110556.
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–27. https://doi.org/10.1056/NEJMoa1002028.
Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–17. https://doi.org/10.1182/blood-2009-04-214957.
Galli S, McLornan D, Harrison C. Safety evaluation of ruxolitinib for treating myelofibrosis. Expert Opin Drug Saf. 2014;13(7):967–76. https://doi.org/10.1517/14740338.2014.916273.
Vannucchi AM. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(17):1670–1. https://doi.org/10.1056/NEJMc1502524.
Mesa R, Vannucchi AM, Yacoub A, Zachee P, Garg M, Lyons R, et al. The efficacy and safety of continued hydroxycarbamide therapy versus switching to ruxolitinib in patients with polycythaemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Br J Haematol. 2017;176(1):76–85. https://doi.org/10.1111/bjh.14382.
Passamonti F, Griesshammer M, Palandri F, Egyed M, Benevolo G, Devos T, et al. Ruxolitinib for the treatment of inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): a randomised, open-label, phase 3b study. Lancet Oncol. 2017;18(1):88–99. https://doi.org/10.1016/S1470-2045(16)30558-7.
Lussana F, Cattaneo M, Rambaldi A, Squizzato A. Ruxolitinib-associated infections: a systematic review and meta-analysis. Am J Hematol. 2018;93(3):339–47. https://doi.org/10.1002/ajh.24976.
Tong LX, Jackson J, Kerstetter J, Worswick SD. Reactivation of herpes simplex virus infection in a patient undergoing ruxolitinib treatment. J Am Acad Dermatol. 2014;70(3):e59–60. https://doi.org/10.1016/j.jaad.2013.09.035.
Caocci G, Murgia F, Podda L, Solinas A, Atzeni S, La Nasa G. Reactivation of hepatitis B virus infection following ruxolitinib treatment in a patient with myelofibrosis. Leukemia. 2014;28(1):225–7. https://doi.org/10.1038/leu.2013.235.
Tsukamoto Y, Kiyasu J, Tsuda M, Ikeda M, Shiratsuchi M, Ogawa Y, et al. Fatal disseminated tuberculosis during treatment with ruxolitinib plus prednisolone in a patient with primary myelofibrosis: a case report and review of the literature. Intern Med (Tokyo, Jpn). 2018;57(9):1297–300. https://doi.org/10.2169/internalmedicine.9165-17.
Palandri F, Tiribelli M, Benevolo G, Tieghi A, Cavazzini F, Breccia M, et al. Efficacy and safety of ruxolitinib in intermediate-1 IPSS risk myelofibrosis patients: results from an independent study. Hematol Oncol. 2018;36(1):285–90. https://doi.org/10.1002/hon.2429.
Polverelli N, Palumbo GA, Binotto G, Abruzzese E, Benevolo G, Bergamaschi M, et al. Epidemiology, outcome, and risk factors for infectious complications in myelofibrosis patients receiving ruxolitinib: a multicenter study on 446 patients. Hematol Oncol. 2018;36:561–9. https://doi.org/10.1002/hon.2509.
Lescuyer S, Ledoux MP, Gravier S, Natarajan-Ame S, Duval C, Maloisel F, et al. Tuberculosis and atypical mycobacterial infections in ruxolitinib-treated patients with primary or secondary myelofibrosis or polycythemia vera. Int J Infect Dis. 2019;80:134–6. https://doi.org/10.1016/j.ijid.2019.01.002.
McLornan DP, Khan AA, Harrison CN. Immunological consequences of JAK inhibition: friend or foe? Curr Hematol Malig Rep. 2015;10(4):370–9.
Chen S, Yan H, Zhang L, Kong W, Sun Y, Zhang W, et al. Cryptococcus neoformans infection and immune cell regulation in human monocytes. Cell Physiol Biochem. 2015;37(2):537–47. https://doi.org/10.1159/000430375.
Moodley D, Yoshida H, Mostafavi S, Asinovski N, Ortiz-Lopez A, Symanowicz P, et al. Network pharmacology of JAK inhibitors. Proc Natl Acad Sci U S A. 2016;113(35):9852–7. https://doi.org/10.1073/pnas.1610253113.
• Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. 2017;18(4):374–84. https://doi.org/10.1038/ni.3691 Important review about the Jak-STAT pathway focusing on immune cell function, human disease, and therapeutic interventions of drugs targeting this pathway.
Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597–612. https://doi.org/10.1016/S0076-6879(05)07047-3.
Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5(10):835–44. https://doi.org/10.1038/nrd2130.
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–109. https://doi.org/10.1158/0008-5472.CAN-04-1443.
Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66(24):11851–8. https://doi.org/10.1158/0008-5472.CAN-06-1377.
Watt TC, Cooper T. Sorafenib as treatment for relapsed or refractory pediatric acute myelogenous leukemia. Pediatr Blood Cancer. 2012;59(4):756–7. https://doi.org/10.1002/pbc.23394.
Sammons SL, Pratz KW, Smith BD, Karp JE, Emadi A. Sorafenib is tolerable and improves clinical outcomes in patients with FLT3-ITD acute myeloid leukemia prior to stem cell transplant and after relapse post-transplant. Am J Hematol. 2014;89(9):936–8. https://doi.org/10.1002/ajh.23782.
Takeda H, Nishikawa H, Iguchi E, Matsuda F, Kita R, Kimura T, et al. Sorafenib-induced acute interstitial pneumonia in patients with advanced hepatocellular carcinoma: report of three cases. Clin J Gastroenterol. 2012;5(4):407–12. https://doi.org/10.1007/s12328-012-0339-9.
Chen CB, Wu MY, Ng CY, Lu CW, Wu J, Kao PH, et al. Severe cutaneous adverse reactions induced by targeted anticancer therapies and immunotherapies. Cancer Manag Res. 2018;10:1259–73. https://doi.org/10.2147/CMAR.S163391.
Macdonald JB, Macdonald B, Golitz LE, LoRusso P, Sekulic A. Cutaneous adverse effects of targeted therapies: part I: inhibitors of the cellular membrane. J Am Acad Dermatol. 2015;72(2):203–18; quiz 19-20. https://doi.org/10.1016/j.jaad.2014.07.032.
Chen KH, Weng MT, Chou YH, Lu YF, Hsieh CH. Epigastric distress caused by esophageal candidiasis in 2 patients who received sorafenib plus radiotherapy for hepatocellular carcinoma: case report. Medicine. 2016;95(11):e3133. https://doi.org/10.1097/MD.0000000000003133.
Coppola R, Zanframundo S, Rinati MV, Carbotti M, Graziano A, Galati G, et al. Rhodotorula mucilaginosa skin infection in a patient treated with sorafenib. J Eur Acad Dermatol Venereol. 2015;29(5):1028–9. https://doi.org/10.1111/jdv.12455.
Dubourdeau M, Athman R, Balloy V, Huerre M, Chignard M, Philpott DJ, et al. Aspergillus fumigatus induces innate immune responses in alveolar macrophages through the MAPK pathway independently of TLR2 and TLR4. J Immunol (Baltimore, Md : 1950). 2006;177(6):3994–4001. https://doi.org/10.4049/jimmunol.177.6.3994.
Jia XM, Tang B, Zhu LL, Liu YH, Zhao XQ, Gorjestani S, et al. CARD9 mediates Dectin-1-induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J Exp Med. 2014;211(11):2307–21. https://doi.org/10.1084/jem.20132349.
Hipp MM, Hilf N, Walter S, Werth D, Brauer KM, Radsak MP, et al. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood. 2008;111(12):5610–20. https://doi.org/10.1182/blood-2007-02-075945.
Martin del Campo SE, Levine KM, Mundy-Bosse BL, Grignol VP, Fairchild ET, Campbell AR, et al. The Raf kinase inhibitor sorafenib inhibits JAK-STAT signal transduction in human immune cells. J Immunol (Baltimore, Md : 1950). 2015;195(5):1995–2005. https://doi.org/10.4049/jimmunol.1400084.
Markham A. Fostamatinib: first global approval. Drugs. 2018;78(9):959–63. https://doi.org/10.1007/s40265-018-0927-1.
McKeage K, Lyseng-Williamson KA. Fostamatinib in chronic immune thrombocytopenia: a profile of its use in the USA. Drugs Ther Perspect. 2018;34(10):451–6. https://doi.org/10.1007/s40267-018-0551-x.
• Drummond RA, Franco LM, Lionakis MS. Human CARD9: a critical molecule of fungal immune surveillance. Front Immunol. 2018;9:1836. https://doi.org/10.3389/fimmu.2018.01836 A review on the important adaptor signaling molecule CARD-9 and the critical role Syk-CARD-9 signaling in human antifungal host defense, underscoring the potentially detrimental effects of Syk inhibitors .
•• Drummond RA, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, et al. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog. 2015;11(12):e1005293. https://doi.org/10.1371/journal.ppat.1005293 This study described for the first time the protective role of CARD9 in orchestrating neutrophil recruitment to the Candida-infected brain. The conclusions drawn from this study are highly relevant in light of the clinical use of fostamatinib and other SMKIs that may block Syk-CARD9-dependent signaling .
•• Drummond RA, Swamydas M, Oikonomou V, Zhai B, Dambuza IM, Schaefer BC, et al. CARD9(+) microglia promote antifungal immunity via IL-1beta- and CXCL1-mediated neutrophil recruitment. Nat Immunol. 2019;20(5):559–70. https://doi.org/10.1038/s41590-019-0377-2 This study provides novel mechanistic insight into how CARD9-signaling in brain-resident microglia regulates protective neutrophil recruitment to the Candida-infected brain.
Drewniak A, Gazendam RP, Tool AT, van Houdt M, Jansen MH, van Hamme JL, et al. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood. 2013;121(13):2385–92. https://doi.org/10.1182/blood-2012-08-450551.
• Rieber N, Gazendam RP, Freeman AF, Hsu AP, Collar AL, Sugui JA, et al. Extrapulmonary Aspergillus infection in patients with CARD9 deficiency. JCI Insight. 2016;1(17):e89890. https://doi.org/10.1172/jci.insight.89890 This study describes CARD9-deficient patients developing extrapulmonary aspergillosis, associated with a defect in neutrophil accumulation in the infected tissue.
LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8(6):630–8. https://doi.org/10.1038/ni1460.
Funding
This work was supported by the Division of Intramural Research (DIR) of the NIAID, NIH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Marissa Zarakas, Jigar Desai, Georgios Chamilos, and Michail Lionakis declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Fungal Genomics and Pathogenesis
Rights and permissions
About this article

Cite this article
Zarakas, M.A., Desai, J.V., Chamilos, G. et al. Fungal Infections with Ibrutinib and Other Small-Molecule Kinase Inhibitors. Curr Fungal Infect Rep 13, 86–98 (2019). https://doi.org/10.1007/s12281-019-00343-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12281-019-00343-9