
Abstract
Purpose of Review
Autoinflammatory diseases (AIDs) constitute several disorders that share similar characteristics, clinical features, disease course, and prognosis. They are characterized by the presence of recurrent episodes of unprovoked inflammation due to dysregulated innate immune system in the absence of autoantibodies or infections. AIDs include periodic fever syndromes and other less commonly growing list of syndromes. In this review, vasculitis associated with different AIDs will be highlighted.
Recent Findings
Vasculitis is inflammation and necrosis of the blood vessels causing impaired blood flow, ischemia, and infarction of the dependent tissues. It is a very rare manifestation of AIDs and when it occurs, the skin is the most affected tissue than any other organs such as kidneys, lungs, or CNS.
Summary
Although vasculitis and AIDs share similar manifestations such as fever, skin rashes, and neuropathy, vasculitis is not a characteristic feature of AIDs and still not clear if it represents a main clinical feature or a manifestation of other disease process.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The term “autoinflammatory diseases” was first described in 1999 to distinguish this group of disorders from other well-known autoimmune diseases [1]. It is characterized by the presence of recurrent episodes of unprovoked inflammation due to dysregulated innate immune system in the absence of autoantibody production or infections. Most patients present in infancy or early childhood and the diagnosis should be suspected in patients with recurrent episodes of self-limited unexplained inflammatory symptoms [2]. AIDs include periodic fever syndromes and other less commonly growing list of syndromes. The periodic fever syndromes include familial Mediterranean fever (FMF), tumor necrosis factor (TNF) receptor-1-associated periodic syndrome (TRAPS), hyperimmunoglobulin D syndrome (HIDS), cryopyrin-associated periodic syndromes (CAPS), and periodic fevers with aphthous stomatitis, pharyngitis, and adenitis (PFAPA).
The spectrum of autoinflammatory diseases is expanding and the most recently recognized disorder is the newly described autosomal dominant autoinflammatory syndrome caused by pathogenic variants of tumor necrosis factor-alpha-induced protein 3 (TNFAIP3), A20 haploinsufficiency (HA20) [3••]. It also extends beyond classic hereditary periodic fever syndromes such as FMF and includes disorders such as Behcet’s disease, which is considered a secondary autoinflammatory disease [4]. Cryopyrin-associated periodic syndrome (CAPS) encompasses a group of disorders including, in order of increasing severity, familial cold autoinflammatory syndrome, Muckle–Wells syndrome, and chronic infantile neurological cutaneous articular (CINCA) syndrome. CAPS are caused by mutation of the gene CIAS1 located on chromosome 1q44 and encodes a protein previously called cryopyrin but is now known as the NOD-like receptor 3 (NLRP3) protein [5].
Vasculitis denotes inflammation and necrosis of the blood vessels causing impaired blood flow, ischemia, and infarction of the dependent tissues. In general, the vessels affected in vasculitis vary in size, type, and location. It may occur as a primary process or secondary to another underlying disease. Although the classifications and nomenclatures are an evolving topic, its classification depends on the blood vessel size, type of organ involved, systemic disease, and probable etiology. Behcet’s disease is the only autoinflammatory disease present in this nomenclature under variable vessel vasculitis which can affect any type of blood vessel [6].
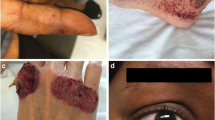
Vasculitis is a very rare manifestation of AIDs and the skin is the most affected than any other organs such as kidneys, lungs, or CNS. Although vasculitis and AIDs share similar manifestations such as fever, skin rashes, and neuropathy, vasculitis is not a characteristic feature of AIDs and it is still not clear if it represents a main clinical feature or only a manifestation of other disease process [7••, 8•]. The most commonly described histopathological manifestation associated with AIDs is cutaneous leukocytoclastic vasculitis [8•].
In this report, the different types of autoinflammatory diseases and the associated vasculitis will be described. Whether the clinical expression of the vasculitis process associated with AIDs differs to that present as separate clinical entity will be explored.
Familial Mediterranean Fever
Familial Mediterranean fever (FMF) is the most common autoinflammatory disease (AID) and characterized by self-limited episodes of recurrent fever which may be accompanied by serositis, arthritis, dermal manifestations, and long-term complications, mainly renal. The MEFV is the gene responsible for familial Mediterranean inheritance that was described in 1997 and encodes for the protein pyrin, an important player in the innate immune system and the component of inflammasome which leads to exaggerated inflammatory response through uncontrolled production of interleukin-1 [9].
Henoch–Schönlein purpura and polyarteritis nodosa (PAN) are the most common vasculitides described in FMF, Table 1 [7••, 9]. Jain et al. have recently described that both vasculitides may infrequently occur in FMF [22]. Ozdogan et al. had previously described the presence of Henoch–Schönlein purpura in 15 patients (7%) and polyarteritis nodosa in two patients (1%) in 207 patients with FMF [23].
Immunoglobulin A vasculitis (IgA vasculitis), IgAV, formerly called Henoch–Schönlein purpura [HSP]), is a small vessel vasculitis and the most common vasculitis in children. It is the most reported vasculitis associated with FMF [22]. IgAV is characterized by a tetrad of palpable purpura due to cutaneous vasculitis, arthralgia/arthritis, abdominal pain, and hematuria due to renal involvement [24]. IgAV was described in 17 out of 470 patients with FMF by Sohar E et al., which was the largest report of FMF to date [25]. Henoch–Schönlein purpura associated with FMF differs from the typical isolated Henoch–Schönlein purpura and should be considered associated rather than co-existing as separate clinical entities. Henoch–Schönlein purpura associated with FMF had a more severe and protracted course with high fever and severe joint pain when compared with those patients who did not have FMF [26].
Polyarteritis nodosa (PAN) is the second most common vasculitis reported in FMF patients and of note, MEFV gene mutation has been detected in PAN without FMF symptoms [27]. It is a systemic necrotizing vasculitis that typically affects medium-sized muscular arteries. When presented in FMF patients, it tends to present earlier in age and patients suffer from severe myalgia compared to the classic PAN without FMF [7••].
Although erysipela-like erythema (ELE), which is a pathognomonic skin manifestation of FMF, is not considered vasculitis, in a study of seven patients with FMF in whom ELE was developed, the histologic examination showed sparse perivascular infiltrate of lymphocytes, neutrophils histiocytes, and nuclear dust, and in the vessel walls, deposits of IgM, C3, and fibrinogen were observed [28].
Protracted febrile myalgia which is considered to be a vasculitic manifestation of FMF is characterized by episodes of fever, myalgia, elevated ESR, and normal CK. It has been recognized as a well-known manifestation of a severe course of FMF and it is associated with homozygosity for M694V mutation [29]. Rare cases of other types of vasculitis co-existing with FMF such as central nervous system vasculitis [30] and coronary vasculitis [31] have been reported.
TNF Receptor-Associated Periodic Syndrome
The tumor necrosis factor receptor-associated periodic fever syndrome (TRAPS) also known as familial Hibernian fever was described in 1982. It is associated with mutations in the gene for tumor necrosis factor receptor superfamily member 1A (TNFRSF1A), on chromosome 12. It is a very rare diagnosis with an estimated prevalence of one per million. It is characterized by attacks of fever in > 83% of patients and accompanied by symptoms including diffuse limb pain, abdominal pain, and rash. The attacks differ from typical FMF in that it can be discrete or near continuous and are often prolonged, lasting several weeks. It is accompanied by a variety of features, including fever, abdominal pain, rash, eye manifestations, headache, pleuritic pain, and lymphadenopathy [32].
Vasculitis has been rarely reported with TRAPS. Reviewing the literature, one case of confirmed TRAPS presented with migratory erythematous macules and skin biopsy showed a small vessel vasculitis and panniculitis [10]. The patient was a 66-year-old white woman who had relapsing episodes of fever, migratory erythematous macules, oligoarthritis, and myalgias for years. The ESR, C reactive protein, and leucocyte counts rose dramatically during her recurrent attacks. ANCA with specificity for human leucocyte elastase (HLE-ANCA) were repeatedly detected, but no other symptoms of systemic vasculitis were found. Analysis of the tumor necrosis factor receptor super family (TNFRSF) 1A gene was performed demonstrating the R92Q mutation. TRAPS was diagnosed, and the patient responded favorably to treatment with the human soluble p75 tumor necrosis factor a (TNF-a) receptor fusion protein etanercept. In conclusion, vasculitis is not a common feature of TRAPS and no other reported cases were found.
Hyper-IgD Syndrome (Mevalonate Kinase Disease)
Hyper-IgD syndrome (HIDS) or Mevalonate kinase disease (MKD) is a rare, autosomal recessive genetic disorder, which typically presents during the first year of life. It results from mutations in the MVK gene, which encodes the enzyme mevalonate kinase (MVK). Loss of the enzyme activity result in both the milder phenotype known as hyperimmunoglobulin D syndrome and the more severe phenotype, mevalonic aciduria. It is characterized by unremitting recurrent fever lasting several days associated with lymphadenopathy, splenomegaly, arthralgia/arthritis, abdominal pain, rash, and an elevated serum polyclonal immunoglobulin D (IgD) level [33].
Mild features of vasculitis, sweet-like features, cellulitis-like findings, and deep vasculitis characteristics were the most common findings in skin biopsies of cutaneous manifestations of 40 patients with the hyper-IgD syndrome. Erythematous macules were the most common cutaneous manifestation, followed by erythematous papules, urticarial lesions, and erythematous nodules [11]. Leukocytoclastic vasculitis has been reported in some case patients in association with HIDS. The most recent case report was a 24-year-old man with a history of episodes of fever, abdominal pain, rash, diarrhea, pleuritis, and lymphadenopathy since age 19 years. He was admitted with fever up to 41 °C and erythematous erysipela-like rash. The skin biopsy revealed leukocytoclastic vasculitis, the IgD level was 150 mg/ml (normal range 0–8 mg/ml), and the gene mutation analysis of MV444K revealed a single V377I mutation in exon 11 with no MEFV or TNF receptor mutations [8•].
Cryopyrin-Associated Periodic Syndromes
Cryopyrin-associated periodic syndromes (CAPS) or cryopyrinopathies are autosomal dominant autoinflammatory disorders in which recurrent inflammation with fever is a manifestation shared by all types of CAPS and joint manifestation is the most common finding [34].
Three clinically overlapping autoinflammatory disorders constitute the cryopyrin-associated periodic syndromes (CAPS) including familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS), and neonatal-onset multisystem inflammatory disorder (NOMID), also known as chronic infantile neurologic cutaneous and articular syndrome (CINCA). It consists of a continuum of autoinflammatory diseases caused by a defect in interleukin-1β regulation. The CAPS gene, NLRP3 (nucleotide-binding domain, leucine-rich repeat family, pyrin domain containing 3), previously named CIAS1 (cold-induced autoinflammatory syndrome 1) was identified in 2001. FCAS is the mildest, NOMID is the most severe, and MWS is in the middle of the spectrum [35]. The cutaneous eruption in CAPS is classically the neutrophilic urticarial dermatosis which is characterized by neutrophilic perivascular and interstitial inflammatory infiltrate [12].
Pyogenic Sterile Arthritis, Pyoderma Gangrenosum, and Acne Syndrome
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome (PAPA) is a rare autosomal dominant autoinflammatory condition that presents in the first decade of life and primarily affecting joints and skin, manifesting as pyogenic arthritis, pyoderma gangrenosum, and acne. Pyoderma gangrenosum pathology consists of vascular changes suggestive of lymphocytic vasculitis. Leukocytoclastic vasculitis may also be present. An association between vasculitis and PAPA syndrome was first described by Khatibi K et al. in a young patient with PAPA who developed an unusual cerebral arterial vasculitis that resulted in subarachnoid hemorrhage from a ruptured dissecting posterior cerebral artery aneurysm. This aneurysm was successfully treated by endovascular coil and no histology or biopsy mentioned [13].
Blau Syndrome
Blau syndrome (BS) and early-onset sarcoidosis are respectively the familial and sporadic forms of granulomatous autoinflammatory disease that results from mutations in or near the nucleotide oligomerization domain of the NOD2 pattern recognition receptor. It is an autosomal dominant condition characterized by granulomatous inflammation of the skin, eye, and joints. Large vessel vasculitis (Takayasu’s-like) and leukocytoclastic vasculitis have been recently reported in association with Blau syndrome [14].
Periodic Fever with Aphthous Stomatitis, Pharyngitis, and Adenitis (PFAPA)
Periodic fever with aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a relatively common disease compared to other autoinflammatory syndromes and considered to be the most common recurrent fever syndrome in children. It generally occurs in children of less than 5 years of age and mainly in the first year of life [36]. It is characterized by periodically recurrent fevers, oropharyngeal inflammation, and adenitis, which recently has been also recognized in adulthood. The etiology and genetic causes are still uncertain [37].
Kawasaki disease (KD) was reported in a patient with PFAPA [15] and the incidence of PFAPA was high in KD patients in a study by Broderick et al. in which they reported four patients with PFAPA among 84 patients with KD and suggested that these patients might have a genetic propensity toward altered immune response and autoinflammatory syndrome. The association between KD and PFAPA may represent a genetic predisposition to dysregulated innate immune response [16].
Behcet’s Disease
Behcet’s disease (BD) is now accepted to be an autoinflammatory disease [4], although not classified under this family of disorders [6]. Most clinical manifestations of Behçet syndrome are believed to be due to vasculitis and are characterized by recurrent and usually painful mucocutaneous ulcers. Most cases are sporadic and affect young adults 20 to 40 years of age and more severe in young, male patients from Middle- or Far-Eastern Asia [38].
Vascular involvement occurred in 12.8% of BD patients in a recent retrospective study of 796 patients. Venous involvement resulting in venous thrombosis is more common than arterial involvement and the most common type of vascular involvement is deep venous thrombosis in lower extremities. Female BD patients were more often involved with arterial lesions, whereas male BD patients developed venous lesions more often than females [39]. The small vessel vasculitis is the most common vasculitis in Behcet’s disease. Large vessel involvement occurs in approximately one third of patients [40].
Stimulator of Interferon Genes (STING)-Associated Vasculopathy with Onset in Infancy
STING-associated vasculopathy with onset in infancy (SAVI) is a newly described autoinflammatory syndrome caused by gain-of-function mutations in TMEM173, the gene that encodes the STING protein. The mutation accounts for constitutive activation of the STING-interferon pathway, leading to overproduction of interferon. The original report included six patients in early infancy with features of cutaneous vasculitis and severe interstitial lung disease in three of them. It presents with systemic inflammation, severe scaling skin lesions which progressed to acral necrosis, gangrenous digits, dystrophic nail changes, and resorption of distal phalanges. The dermal infiltrate showed leukocytoclastic vasculitis and microthrombotic angiopathy of small dermal vessels [17]. Leukocytoclastic vasculitis associated with confirmed SAVI was described in three other cases of AIDs-associated leukocytoclastic vasculitis [8•].
Deficiency of Adenosine Deaminase 2
Deficiency of adenosine deaminase type 2 (DADA2 syndrome) is a newly described autoinflammatory syndrome which was first described in 2014 by two independent groups [18, 19]. It is an autosomal recessive disease resulting from loss-of-function mutations in ADA2, formerly named CECR1 (cat eye syndrome chromosome region, candidate 1) gene. It develops mainly in childhood with 77% presented before the age of 10 years and characterized by recurrent fevers, vascular features, and mild immunodeficiency. Vasculopathy of small- and medium-sized arteries is the major clinical feature of DADA2 and it has a highly variable clinical presentation which renders early diagnosis difficult. It causes polyarteritis nodosa vasculopathy with highly varied clinical expression. Clinical manifestations range from severe or fatal systemic vasculitis or multiple strokes in children to limited cutaneous manifestations in middle-age persons. More recent reports have stressed hematological manifestations with pure red cell aplasia, thrombocytopenia, and neutropenia. ADA1 mutations are a well-known cause of severe combined immune deficiency. However, ADA2 has mild to no apparent immune deficiency. The mainstay of treatment is TNF-inhibition, which is successful in suppressing inflammation and in prevention of vascular events [18,19,20].
A20 Haploinsufficiency
It is a new autoinflammatory disease characterized by high penetrance heterozygous loss-of-function mutations in TNFAIP3 which encodes the NF-ΚB regulatory protein (A20), also known as TNAP3 and plays a crucial role in the negative regulation of inflammation and immunity leading to haploinsufficiency of A20 (HA20). It was initially described by Zhou Q et al. in six unrelated families with early-onset systemic inflammation resembles Behçet’s disease [21•].
Aeschlimann et al. recently described this disorder in the largest cohort to date of 16 patient [3••]. The disease is characterized by childhood-onset systemic inflammation and “Behçet-like” manifestations with recurrent oral, genital and/or gastrointestinal ulcers leading to end-organ damage and death. Two patients with neurological manifestations were diagnosed with central nervous system (CNS) vasculitis (one based on brain imaging and the other one based on a frontal lobe punctate) and one patient was diagnosed with retinal vasculitis. (Table 1).
Conclusion
Vasculitis and vasculopathy rarely occur in AIDs. When present, leukocytoclastic vasculitis, polyarteritis nodosa, and large vessel involvement or “Takayasu-like arteritis” are most commonly seen. Newer AID syndromes are increasingly recognized i.e., DADA2 syndrome in which vascular involvement is an important component and causes early polyarteritis nodosa vasculopathy with highly varied clinical expression. A20 haploinsufficiency is a recently described AID which presents in childhood with systemic inflammation and “Behçet-like” manifestations; CNS vasculitis and retinal vasculitis have been reported.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–44.
Lachmann H. Periodic fever syndromes. Best Pract Res Clin Rheumatol. 2017;31(4):596–609.
•• Aeschlimann FA, Batu ED, Canna SW, Go E, Gül A, Hoffmann P, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis. 2018;77:728–35 This cohort describes the most recent autoinflammatory syndrome, HA20.
Gul A. Behcet’s syndrome is an autoinflammatory syndrome. Curr Drug Targets Inflamm Allergy. 2005;4:81–3.
Lucherini OM, Rigante D, Sota J, Fabiani C, Obici L, et al. Updated overview of molecular pathways involved in the most common monogenic autoinflammatory diseases. Clin Exp Rheumatol. 2018;Suppl 110(1):3–9.
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65(1):1–11.
•• Peleg H, Ben-Chetrit E. Vasculitis in the autoinflammatory diseases. Curr Opin Rheumatol. 2017;29(1):4–11 This review provides more detailed data about vasculitis in autoinflammatory syndromes.
• Ginsberg S, Rosner I, Rozenbaum M, Slobodin G, Zilber K. Autoinflammatory associated vasculitis. Semin Arthritis Rheum. 2016;46(3):367–71 This report also emphasizes the leucocytoclastic vasculitis across spectrum of autoinflammatory diseases.
Alghamdi M. Familial Mediterranean fever, review of the literature. Clin Rheumatol. 2017;36(8):1707–13.
Lamprecht P, Moosig F, Adam-Klages S, Mrowietz U, Csernok E, Kirrstetter M, et al. Small vessel vasculitis and relapsing panniculitis in tumour necrosis factor receptor associated periodic syndrome (TRAPS). Ann Rheum Dis. 2004;63:1518–20.
Drenth JP, Boom BW, Toonstra J, Van der Meer JW. Cutaneous manifestations and histologic findings in the hyperimmunoglobulinemia D syndrome. International hyper IgD study group. Arch Dermatol. 1994;130(1):59–65.
Kolivras A, Theunis A, Ferster D, Lipsker D, et al. Cryopyrin-associated periodic syndrome: an autoinflammatory disease manifested as neutrophilic urticarial dermatosis with additional perieccrine involvement. J Cutan Pathol. 2011;38(2):202–8.
Khatibi K, Heit JJ, Telischak NA, Elbers JM, Do HM. Cerebral vascular findings in PAPA syndrome: cerebral arterial vasculopathy or vasculitis and a posterior cerebral artery dissecting aneurysm. BMJ Case Rep. 2015;24:2015.
Rosé CD, Pans S, Casteels I, Anton J, Bader-Meunier B, et al. Blau syndrome: cross-sectional data from a multicentre study of clinical, radiological and functional outcomes. Rheumatology (Oxford). 2015;54(6):1008–16.
Ninomiya T, Takada H, Nagatomo Y, Nanishi E, Nagata H, Yamamura K, et al. Development of Kawasaki disease in a patient with PFAPA. Pediatr Int. 2013;55(6):801–2.
Broderick L, Tremoulet AH, Burns JC, Bastian JF, Hoffman HM. Recurrent fever syndromes in patients after recovery from Kawasaki syndrome. Pediatrics. 2011;127:e489–93.
Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–18.
Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370(10):911–20.
Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370(10):921–31.
Meyts I, Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. 2018;38(5):569–78.
• Zhou Q, Wang H, Schwartz DM, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. 2016;48:67–73 This study provides the initial report of A20 haploinsufficiency and associated mutations.
Jain A, Misra DP, Sharma A, Wakhlu A, Agarwal V, Negi VS. Vasculitis and vasculitis-like manifestations in monogenic autoinflammatory syndromes. Rheumatol Int. 2018;38(1):13–24.
Ozdogan A, Arisoy N, Kasapcapur O, Sever L, et al. Vasculitis in familial Mediterranean fever. J Rheumatol. 1997;24(2):323–7.
Piram M, Mahr A. Epidemiology of immunoglobulin A vasculitis (Henoch-Schönlein): current state of knowledge. Curr Opin Rheumatol. 2013;25(2):171–8.
Sohar E, Gafni J, Pras M, et al. Familial Mediterranean fever: a survey of 470 cases and review of the literature. Am J Med. 1976;43(2):227–53.
Ben-Chetrit E, Yazici H. Nonthrombocytopenic purpura in familial Mediterranean fever—co-morbidity with Henoch-Schonlein purpura or an additional rare manifestation of familial Mediterranean fever? Rheumatology (Oxford). 2016;55(7):1153–8.
Yalcinkaya F, Ozcakar ZB, Kasapcopur O, Ozturk A, Akar N, Bakkaloglu A, et al. Prevalence of the MEFV gene mutations in childhood polyarteritis nodosa. J Pedi- atr. 2007;151:675–8.
Barzilai A, Langevitz P, Goldberg I, Kopolovic J, Livneh A, Pras M, et al. Erysipelas-like erythema of familial Mediterranean fever: clinicopathologic correlation. J Am Acad Dermatol. 2000;42:791–5.
Langevitz P, Zemer D, Livneh A, Shemer J, Pras M. Protracted febrile myalgia in patients with familial Mediterranean fever. J Rheumatol. 1994;21:1708–9.
Luger S, Harter PN, Mittelbronn M, Wagner M, Foerch. Brain stem infarction associated with familial Mediterranean fever and central nervous system vasculitis. Clin Exp Rheumatol. 2013;31(3):93–5.
Serrano R, Martinez MA, Andres A, Morales JM, Samartin R. Familial Mediterranean fever and acute myocardial infarction secondary to coronary vasculitis. Histopathology. 1998;33(2):163–7.
Lachmann HJ, Papa R, Gerhold K, Obici L, Touitou I, Cantarini L, et al. The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the Eurofever/EUROTRAPS international registry. Ann Rheum Dis. 2014;73(12):2160–7.
Drenth JP, Haagsma CJ, van der Meer JW. Hyperimmunoglobulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients. International Hyper-IgD Study Group. Medicine (Baltimore). 1994;73(3):133.
Houx L, Hachulla E, Kone-Paut I, Quartier P, Touitou I, Guennoc X, et al. Musculoskeletal symptoms in patients with cryopyrin-associated periodic syndromes: a large database study. Arthritis Rheum. 2015;67(11):3027–36.
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29:301–5.
Førsvoll J, Kristoffersen EK, Øymar K. Incidence, clinical characteristics and outcome in Norwegian children with periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome; a population-based study. Acta Paediatr. 2013;102(2):187–92.
Rigante D, Vitale A, Natale MF, Lopalco G, Andreozzi L, Frediani B, et al. A comprehensive comparison between pediatric and adult patients with periodic fever, aphthous stomatitis, pharyngitis, and cervical adenopathy (PFAPA) syndrome. Clin Rheumatol. 2017;36:463–8.
Savey L, Resche-Rigon M, Wechsler B, Comarmond C, Piette J, Cacoub P, et al. Ethnicity and association with disease manifestations and mortality in Behçet's disease. Orphanet J Rare Dis. 2014;9:42.
Fei Y, Li X, Lin S, Song X, Wu Q, Zhu Y, et al. Major vascular involvement in Behçet's disease: a retrospective study of 796 patients. Clin Rheumatol. 2013;32(6):845–52.
Calamia KT, Schirmer M, Melikoglu M. Major vessel involvement in Behçet disease. Curr Opin Rheumatol. 2005;17(1):1–8.
Acknowledgments
The author acknowledges Professor Luis R. Espinoza, who reviewed this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares that there is no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Vasculitis
Rights and permissions
About this article

Cite this article
Alghamdi, M. Autoinflammatory Disease-Associated Vasculitis/Vasculopathy. Curr Rheumatol Rep 20, 87 (2018). https://doi.org/10.1007/s11926-018-0788-3
Published:
DOI: https://doi.org/10.1007/s11926-018-0788-3