
Abstract
The exact pathophysiological mechanisms leading to the activation and sensitization of the trigeminovascular pathway, which in turns results in the migraine attack, are not completely elucidated. It is likely that direct activation by cortical spreading depression, together with dysfunctional central control of pain, plays a major role in the onset and spreading of the migraine attack. This review focuses on recent structural and functional neuroimaging studies that investigated the role of subcortical and cortical structures in modulating nociceptive input in migraine, which outlined the presence of an imbalance between inhibitory and excitatory modulation of pain processing in the disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Migraine is the most common neurological disorder in the developed world, affecting almost 18 % of the population [1] and causing great economic burden and impact on public health. It is widely accepted that migraine results from a primary brain dysfunction that leads to activation and sensitization of the trigeminovascular pain pathway, whereby sensory afferents from the meningeal and cranial vessels through the trigeminal ganglion project on the caudal trigeminal nucleus and C1-C2 dorsal horns. According to this model, first-order nociceptors in the trigeminal ganglion become activated and sensitized, leading to sequential recruitment and central sensitization of spinal and supraspinal nociceptive neurons. The exact pathophysiological brain mechanisms that constitute the primum movens of this cascade of events are still widely debated. Experimental data has demonstrated that cortical spreading depression (CSD) – a slow, self-propagating wave of neuronal and glial depolarization, and the neurophysiological phenomenon most likely involved with the pathophysiology of the migraine aura [2–4] – is able to activate the trigeminovascular pathway [5] and trigger the migraine attack. Factors influencing susceptibility to CSD, however, as well as the sequence of events leading to headache in migraine without aura (MwoA), remain largely unknown [6].
A large body of evidence supports the view that migraine is also characterized by a dysfunctional central control of pain, as suggested by the observation of ictal and interictal abnormalities in subcortical brainstem and diencephalic nuclei involved in pain modulation [7, 8]. Such findings have led to the juxtaposition of two theories on the pathophysiology of the migraine attack: one that hypothesizes that dysfunction of brainstem nuclei and diencephalic nuclei induces abnormal interpretation of normal sensory input, causing normal sensory flow from the meninges to be interpreted as migraine pain; the other that views the brainstem more as a modulator than a generator of migraine pain, theorizing that dysfunction of inhibitory pain modulation may lead to hyperexcitability along the trigeminovascular pain pathway.
Neuroimaging techniques have provided some insight into the complex pathophysiological mechanisms underlying the onset and spreading of migraine headache, enhancing our understanding of the role of subcortical and cortical structures in modulating nociceptive input. This review highlights recent findings at different anatomical levels of the sensory and pain axis that have helped to expand our knowledge of the putative role of pain-modulating circuits in migraine, which points to the presence of an imbalance between inhibitory and excitatory modulation of pain processing in the disease.
Dorsal Brainstem
Positron emission tomography (PET) imaging studies were the first to examine cerebral blood flow changes during spontaneous migraine attacks in brainstem regions likely involved in nociceptive control, including the dorsolateral pons and caudal midbrain [9]. Blood flow changes in the dorsolateral pons during a migraine episode were also reported in subsequent PET studies [10–12], and appeared to persist after controlling pain with sumatriptan, leading to the conclusion that changes in the dorsolateral pons in migraine are not simply the consequence of headache pain. Activation of this brainstem region, however, does not appear to be entirely specific to migraine headache, or even to pain itself, as it has been reported in neuropathic pain and in conditions characterized by autonomic dysfunction [13–15]. Additionally, controversies exist as to the exact location of brainstem changes identified by first PET observations [16]. Spatial localization of neuronal structures in the brainstem by PET is challenging, and in general there is a discrepancy between the size of the activated clusters in PET studies and the actual size of the nucleus. Major factors that account for this discrepancy include: 1) spatial smoothing of the data, which is usually performed to increase the signal-to-noise ratio and improve comparisons across subjects and conditions; 2) group averaging – nucleus location can show small variation across subjects; and 3) PET contrast itself originates from cerebral blood flow changes and does not measure neuronal activity directly, but rather changes in blood flow following neuronal activity.
By overlapping the original PET activation map of the dorsolateral pons on a brainstem atlas, Borsook and Burstein [16] recently suggested that the area of blood flow changes seemed to correspond anatomically to the location of different brainstem nuclei, including the rostral trigeminal nuclear complex, reticular nucleus, locus coeruleus, cuneiform nucleus, and the inferior colliculus. While the individual role of some of these nuclei in migraine is still uncertain, it is well known that cuneiform nucleus is part of a functional circuit involved in descending pain modulation. It has been suggested that impairment of the descending modulatory pain pathway might be a predefined condition to the occurrence of a migraine attack, by decreasing the threshold of nociceptive inputs from the trigeminocervical complex (TCC). A functional MRI study that examined interictal changes in the brainstem in response to experimental pain stimuli found decreased activity in the cuneiform nucleus of migraineurs relative to controls [17], suggesting that dysfunction of brainstem descending pain modulation may occur even between attacks. Altered activity in the cuneiform nucleus, however, can also be observed during anticipation of pain [18].
Additional evidence of interictal dysfunction in dorsolateral nuclei in the pons can be found in a recent MRI spectroscopy (MRS) study that investigated changes in brainstem metabolites in patients with chronic and episodic migraine [19]. This imaging technique, based on proton resonance (1H-SMR) in predefined regions of interest, is able to provide quantitative information on levels of brain metabolites including N-acetylaspartate (NAA), choline (Cho), and creatine (Cr). A higher NAA/Cr ratio in bilateral rostral dorsal pons was found in patients with episodic migraine compared to patients with chronic migraine and controls. The change in Naa/Cr tended to disappear with disease evolution, as Naa/Cr levels decrease as headache frequency increases in chronic migraine. Based on the knowledge that the Naa/Cr ratio is thought to reflect neuronal content, the authors hypothesized that, in episodic migraine, neurons in the dorsal pons become hypertrophic due to the repetitive activations during migraine attacks. A similar mechanism has been postulated to underlie the observation of increased cortical thickness in episodic migraine [20, 21]. The interpretation of the increase of Naa/Cr ratio in the dorsolateral pons in episodic migraine, however, remains uncertain. Studies from other neurological diseases such as multiple sclerosis show that Naa/Cr ratio also reflects axonal integrity, suggesting that the findings might also underlie microstructural changes in fiber integrity [22].
The Periaqueductal Gray
The periaqueductal gray (PAG), a gray matter structure located around the cerebral aqueduct and organized in four columns, is a known modulator of somatic pain transmission, and is also involved in various physiological processes, including behavioral and emotion control.
A prevailing theory in the pathogenesis of migraine headache is that hyperexcitability develops along the trigeminovascular pathway, likely facilitated by a dysfunction of the descending pain modulatory circuits [7]. The PAG has long been thought to be one of the key regions of the descending pain modulatory system involved in the pathogenesis of migraine, based on early reports of subjects without headache who developed migraine-like episodes after stereotactic placement of electrodes in this area of the brainstem [23].
Previous studies have reported conflicting results relative to the presence of structural abnormalities in the PAG of migraineurs [19, 24, 25]. Recently, data using blood oxygenation level-dependent (BOLD) resting-state functional MRI (rs-fMRI), a technique that allows identification of correlations during rest among remote brain areas (functional connectivity) through their highly correlated low-frequency spontaneous fluctuations, investigated connectivity between the PAG and a subset of brain areas involved in processing and modulation of somatosensory and pain signals in interictal migraine with aura (MwA) and MwoA [26]. Dysfunctional dynamics within PAG networks were observed at different levels. Migraineurs exhibited an increase in rs-fMRI connectivity between the PAG and both nociceptive and sensory processing pathways relative to age- and gender-matched healthy individuals. Connectivity in some of these pathways was stronger as the monthly frequency of migraine attacks increased. Conversely, the greater the number of attacks, the lower the functional rs-fMRI connectivity appeared to be between the PAG and several brain regions with a predominant role in pain modulation, including the prefrontal cortex, anterior cingulate, and amygdala [26]. These findings suggest the presence of an impairment of descending pain modulatory circuits in migraine, which likely leads to loss of pain inhibition and hyperexcitability primarily in nociceptive areas.
A dysfunction in descending pain inhibition in the brainstem has also been hypothesized to contribute to the development of allodynia during migraine attack. Cutaneous allodynia, which is pain to normal innocuous stimuli that develops in about two-thirds of patients during the headache phase, results from sensitization of central neurons along the trigeminovascular pathway [27]. Speculation exists as to whether central sensitization can be indirectly activated through pain modulatory neurons in the brainstem. Data on rs-fMRI intrinsic connectivity of PAG networks found reduced connectivity between PAG, prefrontal regions, and anterior cingulate in migraineurs with a history of allodynia relative to migraineurs without allodynia, highlighting a possible dysfunction of pain modulation [26].
Interestingly, a longitudinal study recently demonstrated that cutaneous allodynia was an independent predictor of migraine chronification, as expressed by increased frequency of migraine attacks [28••]. This finding has been interpreted to reflect functional and/or structural impairment at the level of PAG modulatory networks possibly consequent to repetitive activations of trigeminovascular neurons, which in turn leads to repetitive activations of modulatory pathways involving the PAG.
The Thalamus
Functional and structural thalamic changes in the course of migraine can affect nociception and can contribute to an imbalance between inhibitory and excitatory mechanisms of pain modulation.
Posterior nuclei of thalamus receive direct projections from the trigeminovascular pathway and act as a filter for the somatosensory and nociceptive information [29]. Receptors of the calcitonin gene-related peptide (CGRP) have been described in the ventroposteromedial thalamic nucleus, and antagonist of CGRP can inhibit nociceptive transmission at the level of third-order neurons [30].
Burstein and collaborators identified a population of trigeminovascular neurons in the posterior and lateral posterior thalamic nuclei that may be involved in the perception of whole-body allodynia (abnormal skin sensitivity) and photophobia (abnormal sensitivity to light) during migraine. Using fMRI, they showed that innocuous brush and heat stimulation at the skin of the dorsum of the hand produced larger BOLD responses in the posterior thalamus of subjects undergoing a migraine attack with extracephalic allodynia than corresponding responses registered when the same patients were free of migraine and allodynia [31]. These findings, together with the observed sensitization of sensory neurons in the posterior thalamus in rats, highlight the crucial role of thalamic nuclei in processing nociceptive information along with sensory information from the skin beyond the location of the headache.
An involvement of thalamic pain modulatory regions was recently demonstrated in chronic migraine patients using rs-fMRI [32]. Relative to controls, migraineurs exhibited abnormal functional connectivity between the limbic circuit, the pulvinar and mediodorsal thalamus, and the PAG. Increase in functional connectivity between the insula and the mediodorsal thalamus correlated with the disease duration. A univocal interpretation for such findings is elusive, as changes in rs-fMRI connectivity could reflect either a functional compensatory mechanism to brain structural damage and/or repetitive migraine episodes, or an abnormal functional pattern leading to an aberrant processing of the nociceptive input.
Thalamic structural abnormalities have been identified in migraineurs, particularly in MwA. A structural MRI study found shorter T1 and T2 relaxation times in the thalamus of patients with MwA relative to controls and patients with MwoA [33•]. This has been hypothesized to reflect a higher iron deposit in thalamus of MwA as a consequence of free radical damage and hyperemia during migraine attacks. Magnetization transfer ratio (MTR) [33•], which reflects macromolecule content, was also increased in MwA compared with MwoA patients. Iron deposit in other deep brain nuclei such as the putamen, the globus pallidus, and the red nucleus was observed in a population of young patients with migraine as compared to controls [34], and seemed to increase with the duration of the disease. Taken together, findings from these studies suggest that changes in iron content are more likely a consequence of repeated migraine episodes than a primary cause of migraine headache.
The Hypothalamus
The hypothalamus is involved not only in pain regulation, but also in many vegetative functions, including circadian rhythm, thirst, hunger, and mood control. Several arguments support the involvement of this structure in the physiopathology of migraine [35]. First, its role in pain control has been demonstrated in deep brain stimulation in animal models [36], and several peptides, such as opioid peptides, oxytocin, or somatostatin, have antinociceptive effects [37]. Another argument is based on the higher prevalence of migraine in women, as well as the knowledge of the relationship between hormonal status and migraine frequency, all of which may be linked to luteinizing hormone-releasing hormone (LHRH) secretion. Additionally, premonitory symptoms experienced a few hours before onset of migraine may be a consequence of hypothalamic dysfunction.
Projections from the paraventricular nucleus (PVN) of the hypothalamus may have a role in modulating nociception in migraine. A recent study traced the projections from PVN neurons to the superior salivatory nucleus and medullary trigeminovascular (Sp5C) neurons in rats [38]. Using microinjections of GABAA agonist in the PVN neurons, the authors found an inhibition in both basal and meningeal-evoked activities of Sp5C neurons.
Speculation exists as to whether forebrain/hypothalamic projections to Po and LP thalamic nuclei may play a role in migraine attacks triggered by conditions such as disrupted sleep, hunger, and emotional distress. With the aim of identifying subcortical areas that are in position to directly regulate the activity of thalamic trigeminovascular neurons, Kagan et al. [39] recently mapped anatomically neuronal projections to the Po/LP thalamic region of the rat using injections of the retrograde tracer Fluoro-Gold. Such injections yielded retrogradely labeled neurons in the nucleus of the diagonal band of Broca’s area, the dopaminergic cell groups A11/A13, and the ventromedial and ventral tuberomammillary nuclei of the hypothalamus.
In humans, hypothalamic activation has been observed during the headache attack using PET, and similar to other brainstem structures, has been shown to persist after pain relief by sumatriptan [40], suggesting that hypothalamus activation may occur early, before the onset of migraine attack. Interestingly, in interictal migraine, a positive correlation was recently reported between the number of monthly migraine episodes and resting state correlations between the PAG and the hypothalamus [26], extending previous PET findings and further supporting a role of this subcortical structure in the pathogenesis of migraine. It is also possible that hypothalamic changes in migraine reflect a stress/anxiety response of the brain to worsening of the disease. Further studies are needed to better understand hypothalamic involvement as migraine trigger and/or pain modulator and the link between these processes and mood regulation.
The Amygdala
The amygdala is an essential part of the limbic system, and has many projections to the hypothalamus, thalamus, and insula [41]. While its involvement in fear-feeling, emotion processing, and interoception is well known, it has recently been revealed that the amygdala may play a more important role in migraine than previously thought.
Using fMRI, Stankewitz et al. [42] showed that during a migraine attack following olfactory stimulation, migraineurs had higher activation than controls in the amygdala, insula, dorsal pons, and temporal lobe, suggesting sensitization of these brain structures. No differences were found interictally, however, between controls and patients.
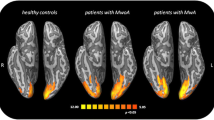
Hadjikhani et al. [43••], using rs-fMRI acquired interictally, demonstrated increased connectivity between the amygdala and visceroceptive regions including insula, thalamus, and secondary somatosensory cortex in MwA and MwoA. Interestingly, this pattern of changes was not detected in other chronic pain conditions, including episodic trigeminal neuralgia and carpal tunnel syndrome. Based on experimental data in the rodent model, which demonstrated that a single episode of CSD (unlikely to elicit pain) induced c-fos expression in the amygdala and behavioral responses consistent with amygdala activation [44], Hadjikhani et al. hypothesized that CSD-induced amygdala activation during repetitive episodes of MwA and MwoA consolidates the connectivity within the visceroceptive cortex, and that amygdala interactions with regions involved in interoception may therefore represent the “missing link” between CSD and the development of migraine headache in humans.
Amygdala connectivity findings further support the concept that migraine is associated with a dysfunctional neurolimbic pain network [45], although it remains to be determined whether and how emotional and/or cognitive and mood influences affect pain perception in migraine. It is known from animal studies that the anterior insula is connected to brainstem structures such as PAG, rostroventromedial medulla (RVM), NCF, and parabrachial nucleus [46], which provides a mechanistic basis to partially explain how emotions and mood might influence changes in the perception of pain intensity. Additionally, as several brainstem descending modulatory regions are either ascending homeostatic integration sites or descending autonomic premotor sites, it is possible that a specific link exists among pain, homeostasis, and interoception.
Higher-Order Cortical Regions
Functional and structural abnormalities in a number of cortical regions involved in somatosensory and pain processing as well as pain modulation have been noted in migraine patients, both during the headache phase and outside episodes of pain [47]. Interictal alterations in cortical processing include changes in cortical excitability in primary sensory regions [48, 49], lack of habituation [50, 51], increased sensitivity to sensory input [52], and abnormalities along pain modulatory cortical networks [26], all of which have contributed to the concept that dysexcitability characterizes the interictal migraineous cortex.
Based on experimental findings in rats demonstrating that trigeminovascular input from the dura distributed nociceptive information to a number of cortical regions, including the primary somatosensory cortex and insula [53], it has been suggested that in some regions, cortical dysexcitability in migraine is, at least in part, the consequence of activation of the trigeminovascular pathway. In this context, cortical abnormalities are seen primarily as the consequence of repetitive episodes of pain. Some authors, however, have suggested that the observed alterations in the cortex may also result from recurrent episodes of CSD which, in migraine without aura, has been hypothesized to occur in clinically silent cortical regions outside the visual cortex.
The knowledge of the relation between functional and structural abnormalities in somatosensory and pain processing regions may aid in understanding the dynamics that occur at multiple cortical levels in migraine. Maleki et al. [54] recently co-localized morphometric and functional abnormalities in cortical regions involved in sensory processing interictally in episodic migraine. By comparing migraineurs with high-frequency (HF) headache episodes relative to patients with low-frequency (LF) migraine, they were able to show higher cortical thickness in the area representing the face in the post-central gyrus in HF, which correlated with stronger functional activation during heat pain stimulation, suggesting adaptation to repeated sensory drive. These findings align with previous observations of a thickening of the somatosensory cortex interictally in patients with MwA and MwoA [20]. The authors also found reduced volume in the cingulate cortex that correlated with lower activation in the HF group during intermittent heat pain stimulation, and similar significant structural and functional differences (HF > LF) in the insula, leading them to speculate that there is a differential response pattern in migraineurs in the sensory versus affective processing regions in the brain. Specifically, they postulated that, in migraine, the brain “reacts” to the increased frequency of migraine attacks by redirecting its resources primarily to regions processing sensory input, reducing the size and activity of the affective processing regions.
In episodic migraine, cortical dysexcitability, expressed as ineffective recruitment of prefrontal inhibitory pathways, may play a role in decreased habituation and enhanced anticipation and attention to pain and other external sensory stimuli. Analysis of cortical responses to predicted and unpredicted noxious heat stimuli in migraineurs by standardized low-resolution electromagnetic tomography key revealed lack of habituation to repeated predicted pain associated with significantly increased pain-evoked potentials amplitude in MwA compared to MwoA and healthy controls [55•]. Relative to controls, however, both MwA and MwoA exhibited enhanced pain-evoked potentials to unpredicted pain stimuli. Using source localization, it was found that deficient adaptive responses to anticipated pain in migraineurs were associated with decreased activation of the anterior-medial prefrontal cortices and subsequent increased somatosensory activity. Additionally, in MwA, the prefrontal-somatosensory dysfunction positively correlated with time since disease onset and concern of upcoming episodes of migraine headache; in MwoA, it increased with higher frequency of migraine. Taken together, these findings suggest that ineffective recruitment of prefrontal inhibitory pathways, accompanied by increased somatosensory activity, on one hand can express the progressive character of the disease, and on the other can be considered as an outcome of patients’ cognitive status interictally.
We are only beginning to disentangle the roles of specific prefrontal and frontal cortical regions in pain perception from other areas of cognitive neuroscience that reflect cognitive, emotional, and interoceptive components of pain conditions [56]. As migraine is known to be associated with mood changes [57], future studies will assess the role of these various components in relation to pain processes.
Conclusion
Our current understanding of migraine has expanded the well-accepted concept of central sensitization along the trigeminovascular pathway to include the role of subcortical-driven cortical hyperexcitability in the pathogenesis of the disease. Hyperexcitability of the migraine brain may result from impaired inhibition or increased facilitation. The consistent observation across studies that changes in brain excitability are frequently associated with disease severity and chronification of migraine suggests that such changes, at least in part, may be expressions of “adaptive” or “maladaptive” mechanisms consequent to repetitive episodes of migraine [58]. In this context, longitudinal studies and comparisons with other chronic pain conditions may help to clarify whether changes in brain excitability are a signature of migraine or represent a common pathway in the chronification of pain across different conditions.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Merikangas KR. Contributions of epidemiology to our understanding of migraine. Headache. 2013;53:230–46.
Hadjikhani N, Sanchez Del Rio M, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687–92.
Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab : Off J Int Soc Cereb Blood Flow Metab. 2011;31:17–35.
Schwedt TJ, Goadsby PJ. 14th International Headache Congress: basic science highlights. Headache. 2010;50:520–6.
Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–42.
Vincent MB, Hadjikhani N. Migraine aura and related phenomena: beyond scotomata and scintillations. Cephalalgia : Int J Headache. 2007;27:1368–77.
Vecchia D, Pietrobon D. Migraine: a disorder of brain excitatory-inhibitory balance? Trends Neurosci. 2012;35:507–20.
Akerman S, Holland PR, Goadsby PJ. Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci. 2011;12:570–84.
Weiller C, May A, Limmroth V, et al. Brain stem activation in spontaneous human migraine attacks. Nat Med. 1995;1:658–60.
Bahra A, Matharu MS, Buchel C, Frackowiak RS, Goadsby PJ. Brainstem activation specific to migraine headache. Lancet. 2001;357:1016–7.
Afridi SK, Giffin NJ, Kaube H, et al. A positron emission tomographic study in spontaneous migraine. Arch Neurol. 2005;62:1270–5.
Afridi SK, Matharu MS, Lee L, et al. A PET study exploring the laterality of brainstem activation in migraine using glyceryl trinitrate. Brain. 2005;128:932–9.
Naliboff BD, Berman S, Chang L, et al. Sex-related differences in IBS patients: central processing of visceral stimuli. Gastroenterology. 2003;124:1738–47.
Dunckley P, Wise RG, Fairhurst M, et al. A comparison of visceral and somatic pain processing in the human brainstem using functional magnetic resonance imaging. J Neurosci : Off J Soc Neurosci. 2005;25:7333–41.
Barman SM, Gebber GL, Kitchens H. Rostral dorsolateral pontine neurons with sympathetic nerve-related activity. Am J Physiol. 1999;276:H401–12.
Borsook D, Burstein R. The enigma of the dorsolateral pons as a migraine generator. Cephalalgia. 2012;32:803–12.
Moulton EA, Burstein R, Tully S, Hargreaves R, Becerra L, Borsook D. Interictal dysfunction of a brainstem descending modulatory center in migraine patients. PLoS One. 2008;3:e3799.
Wager TD, Scott DJ, Zubieta JK. Placebo effects on human mu-opioid activity during pain. Proc Natl Acad Sci U S A. 2007;104:11056–61.
Lai TH, Fuh JL, Lirng JF, Lin CP, Wang SJ. Brainstem 1H-MR spectroscopy in episodic and chronic migraine. J Headache Pain. 2012;13:645–51.
DaSilva AF, Granziera C, Snyder J, Hadjikhani N. Thickening in the somatosensory cortex of patients with migraine. Neurology. 2007;69:1990–5.
Granziera C, DaSilva AF, Snyder J, Tuch DS, Hadjikhani N. Anatomical alterations of the visual motion processing network in migraine with and without aura. PLoS Med. 2006;3:e402.
De Stefano N, Matthews PM, Narayanan S, Francis GS, Antel JP, Arnold DL. Axonal dysfunction and disability in a relapse of multiple sclerosis: longitudinal study of a patient. Neurology. 1997;49:1138–41.
Veloso F, Kumar K, Toth C. Headache secondary to deep brain implantation. Headache. 1998;38:507–15.
Rocca MA, Ceccarelli A, Falini A, et al. Brain gray matter changes in migraine patients with T2-visible lesions: a 3-T MRI study. Stroke; J Cereb Circ. 2006;37:1765–70.
DaSilva AF, Granziera C, Tuch DS, Snyder J, Vincent M, Hadjikhani N. Interictal alterations of the trigeminal somatosensory pathway and periaqueductal gray matter in migraine. Neuroreport. 2007;18:301–5.
Mainero C, Boshyan J, Hadjikhani N. Altered functional magnetic resonance imaging resting-state connectivity in periaqueductal gray networks in migraine. Ann Neurol. 2011;70:838–45.
Burstein R, Cutrer MF, Yarnitsky D. The development of cutaneous allodynia during a migraine attack clinical evidence for the sequential recruitment of spinal and supraspinal nociceptive neurons in migraine. Brain : J Neurol. 2000;123(Pt 8):1703–9.
Louter MA, Bosker JE, van Oosterhout WP, et al. Cutaneous allodynia as a predictor of migraine chronification. Brain : J Neurol. 2013. This longitudinal study demonstrates that cutaneous allodynia, a common feature accompanying migraine attacks and a clinical marker of central sensitization, is an independent predictor of migraine chronification. This finding has been interpreted to reflect functional and/or structural impairment at the level of brainstem pain modulatory networks, possibly consequent to repetitive activations of trigeminovascular neurons, in turn leading to repetitive activations of brainstem modulatory pathways.
Cliffer KD, Burstein R, Giesler Jr GJ. Distributions of spinothalamic, spinohypothalamic, and spinotelencephalic fibers revealed by anterograde transport of PHA-L in rats. J Neurosci. 1991;11:852–68.
Summ O, Charbit AR, Andreou AP, Goadsby PJ. Modulation of nocioceptive transmission with calcitonin gene-related peptide receptor antagonists in the thalamus. Brain. 2010;133:2540–8.
Burstein R, Jakubowski M, Garcia-Nicas E, et al. Thalamic sensitization transforms localized pain into widespread allodynia. Ann Neurol. 2010;68:81–91.
Schwedt TJ, Schlaggar BL, Mar S, et al. Atypical resting-state functional connectivity of affective pain regions in chronic migraine. Headache. 2013;53:737–51.
Granziera C, Daducci A, Romascano D, et al. Structural abnormalities in the thalamus of migraineurs with aura: A multiparametric study at 3 T. Hum Brain Mapp. 2013. This is a multiparametric MRI study that demonstrates broad microstructural alterations in the thalamus of MWA patients that may underlie abnormal cortical excitability control, leading to cortical spreading depression and visual aura.
Kruit MC, Launer LJ, Overbosch J, van Buchem MA, Ferrari MD. Iron accumulation in deep brain nuclei in migraine: a population-based magnetic resonance imaging study. Cephalalgia. 2009;29:351–9.
Alstadhaug KB. Migraine and the hypothalamus. Cephalalgia. 2009;29:809–17.
Carstens E. Hypothalamic inhibition of rat dorsal horn neuronal responses to noxious skin heating. Pain. 1986;25:95–107.
Bartsch T, Levy MJ, Knight YE, Goadsby PJ. Inhibition of nociceptive dural input in the trigeminal nucleus caudalis by somatostatin receptor blockade in the posterior hypothalamus. Pain. 2005;117:30–9.
Robert C, Bourgeais L, Arreto CD, et al. Paraventricular hypothalamic regulation of trigeminovascular mechanisms involved in headaches. J Neurosci. 2013;33:8827–40.
Kagan R, Kainz V, Burstein R, Noseda R. Hypothalamic and basal ganglia projections to the posterior thalamus: Possible role in modulation of migraine headache and photophobia. Neuroscience. 2013;248C:359–68.
Denuelle M, Fabre N, Payoux P, Chollet F, Geraud G. Hypothalamic activation in spontaneous migraine attacks. Headache. 2007;47:1418–26.
Shi CJ, Cassell MD. Cortical, thalamic, and amygdaloid connections of the anterior and posterior insular cortices. J Comp Neurol. 1998;399:440–68.
Stankewitz A, May A. Increased limbic and brainstem activity during migraine attacks following olfactory stimulation. Neurology. 2011;77:476–82.
Hadjikhani N, Ward N, Boshyan J, et al. The missing link: Enhanced functional connectivity between amygdala and visceroceptive cortex in migraine. Cephalalgia : Int J Headache. 2013;33:1264–8. Using rs-fMRI, Hadjikhani et al. demonstrated that functional connectivity is increased in the neurolimbic pain network of Mwa and MwoA but not in other chronic pain conditions. Cortical spreading depression may be driving hyperconnectivity of the amygdala in both types of migraine.
Akcali D, Sayin A, Sara Y, Bolay H. Does single cortical spreading depression elicit pain behaviour in freely moving rats? Cephalalgia : Int J Headache. 2010;30:1195–206.
Maizels M, Aurora S, Heinricher M. Beyond neurovascular: migraine as a dysfunctional neurolimbic pain network. Headache. 2012;52:1553–65.
Sato F, Akhter F, Haque T, et al. Projections from the insular cortex to pain-receptive trigeminal caudal subnucleus (medullary dorsal horn) and other lower brainstem areas in rats. Neuroscience. 2013;233:9–27.
May A. New insights into headache: an update on functional and structural imaging findings. Nat Rev Neurol. 2009;5:199–209.
Coppola G, Curra A, Di Lorenzo C, et al. Abnormal cortical responses to somatosensory stimulation in medication-overuse headache. BMC Neurol. 2010;10:126.
Chen WT, Lin YY, Fuh JL, Hamalainen MS, Ko YC, Wang SJ. Sustained visual cortex hyperexcitability in migraine with persistent visual aura. Brain : J Nneurol. 2011;134:2387–95.
Ozkul Y, Uckardes A. Median nerve somatosensory evoked potentials in migraine. Eur J Neurol : Off J Eur Fed Neurol Soc. 2002;9:227–32.
Schoenen J. Neurophysiological features of the migrainous brain. Neurol Sci : Off J Ital Neurol Soc Ital Soc Clin Neurophysiol. 2006;27 Suppl 2:S77–81.
Boulloche N, Denuelle M, Payoux P, Fabre N, Trotter Y, Geraud G. Photophobia in migraine: an interictal PET study of cortical hyperexcitability and its modulation by pain. J Neurol Neurosurg Psychiatry. 2010;81:978–84.
Noseda R, Constandil L, Bourgeais L, Chalus M, Villanueva L. Changes of meningeal excitability mediated by corticotrigeminal networks: a link for the endogenous modulation of migraine pain. J Neurosci : Off J Soc Neurosci. 2010;30:14420–9.
Maleki N, Becerra L, Brawn J, Bigal M, Burstein R, Borsook D. Concurrent functional and structural cortical alterations in migraine. Cephalalgia : Int J Headache. 2012;32:607–20.
Lev R, Granovsky Y, Yarnitsky D. Enhanced pain expectation in migraine: EEG-based evidence for impaired prefrontal function. Headache. 2013;53:1054–70. This study shows that in both episodic MwA and MwoA, cortical dysexcitability, expressed as ineffective recruitment of prefrontal inhibitory pathways, may play a role in decreased habituation and enhanced anticipation and attention to pain and other external sensory stimuli.
Tracey I, Mantyh PW. The cerebral signature for pain perception and its modulation. Neuron. 2007;55:377–91.
Breslau N, Andreski P. Migraine, personality, and psychiatric comorbidity. Headache. 1995;35:382–6.
Borsook D, Maleki N, Becerra L, McEwen B. Understanding migraine through the lens of maladaptive stress responses: a model disease of allostatic load. Neuron. 2012;73:219–34.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Dr. Caterina Mainero and Dr. Celine Louapre each declare no potential conflict of interest relevant to this article.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Imaging
Rights and permissions
About this article

Cite this article
Mainero, C., Louapre, C. Migraine and Inhibitory System – I Can't Hold It!. Curr Pain Headache Rep 18, 426 (2014). https://doi.org/10.1007/s11916-014-0426-3
Published:
DOI: https://doi.org/10.1007/s11916-014-0426-3