
Abstract
Purpose of Review
The ebb and flow of genetic influence relative to the understanding of craniofacial and dental disorders has evolved into a tacit acceptance of the current genetic paradigm. This review explores the science behind craniofacial and dental disorders through the lens of recent past and current findings and using tooth agenesis as a model of advances in craniofacial genetics.
Recent Findings
Contemporary studies of craniofacial biology takes advantage of the technological resources stemming from the genomic and post-genomic eras. Emerging data highlights the role of key genes and the epigenetic landscape controlling these genes, in causing dentofacial abnormalities. We also report here a novel Glu78FS MSX1 mutation in one family segregating an autosomal dominant form of severe tooth agenesis as an illustration of an evolving theme, i.e., different mutations in the same gene can result in a spectrum of dentofacial phenotypic severity.
Summary
The future of clinical therapeutics will benefit from advances in genetics and molecular biology that refine the genotype-phenotype correlation. Indeed, the past century suggests a continued convergence of genetic science in the practice of clinical dentistry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The mammalian craniofacial region is unique for its breadth of function, ranging from housing the brain and vital sensory organs (optic, auditory, and olfactory) to oronasal functions of respiration, phonation, mastication, and deglutination. Equally unique is its ontogenic and developmental complexity, wherein the neural crest cells represent a central evolutionary feature, responsible for the distinctiveness of the vertebrate head [1, 2]. It is therefore not surprising that a large number of birth disorders involve the craniofacial region and pose considerable challenges to patients as well as clinicians [3]. From a lay point of view, it is apparent that the face represents perhaps the most characteristic human feature with an impact on our physical and social existence. Even more obvious is the similarity of human faces among relatives—past and present—validating the genetic underpinnings of human craniofacial development. Nonetheless, the goal of unraveling the etiology of craniofacial disorders, specifically the contribution of specific genes versus the environment, continues to fuel the debate of “nature versus nurture” and influence the conventional diagnosis and treatment of dental disorders.
Genes Versus Environment
The all-important goal of determining the appropriate diagnosis and treatment modalities for dentofacial disorders, at least partly, relies on dissecting the effect of genes from the environment. Recently, advances in gene sequencing and data mining have provided the field of craniofacial genetics with a better understanding of the molecular fabric that represents our genetic make-up and how this leads to the shape, form, and therefore function of the human face. It is still useful, however, to consider those historical keystones that gave rise to the “nature versus nurture” question so often debated in craniofacial biology and malocclusion. Early studies of the genetic basis of malocclusion sought to determine the causality of specific malocclusions—often using twins. Genetics and heredity, as it relates to malocclusion, was not prominent until the mid-twentieth century, long after the introduction of the laws of segregation by Gregor Mendel (reviewed by Carlson [4]). Nonetheless, in orthodontics, specifically, one can observe that the literature reflects an emerging genetic paradigm proximal to the discovery of the DNA structure by Watson and Crick. Subsequently, in the late twentieth century, heritability and twin studies formed the foundation of a changing landscape for understanding the biology of dental disorders with genetics at the forefront [5,6,7,8,9,10].
The studies of craniofacial and dental traits of the past were pivotal in the development of our theoretical knowledge base of dental disorders but these studies were also limited in some respects (reviewed by Carlson et al. [4]). Today, however, genetic advances within the larger scientific community have benefitted our understanding of craniofacial disorders and in some cases confirmed the speculations of past researchers. The scientific community has witnessed a breakthrough in genotyping technology, especially since the completion of the human genome sequence in 2001 [11]. The human genome contains approximately 3 billion base pairs within 23 pairs of chromosomes. Completion of the entire human genome sequence led to the discovery of over 1500 genes—mainly those responsible for monogenic diseases [12]. The emergence of this genetic paradigm led to rapidly evolving technologies to expand our knowledge beyond monogenic disorders to include complex traits due to more subtle genetic variation. The HapMap (Haplotype Map) Project was one of the first endeavors initiated to unravel the genetic variation using single nucleotide polymorphisms (SNPs). Today, the evolution of molecular biology has led to even more technologies, such as genome-wide association studies (GWAS) and studies epigenetic regulation.
The Impact of Personalized Medicine on Craniofacial Disorders
The end result of this technological boom defined as the “genomic era” was a push toward a translational model in the clinical management of disease. In fact, some investigators and even lay persons might posit that the translational application of genetic information to the clinical diagnosis and treatment was the primary goal of the genomic era. In other words, the genomic era gave birth to the possibility of developing and employing diagnostic tests based on genomics, proteomics, and metabolomics toward predicting the patients’ response to treatment, but served a critical purpose to expand our basic scientific knowledge through unraveling the genome. The resultant field, termed “personalized medicine,” and now dentistry, combines human genome, information technology, and biotechnology with nanotechnology to provide treatment based on individual variation versus population trends [13]. The application of genetic knowledge to craniofacial biology has shaped the current differential diagnosis of disorders in this field ranging from skeletal, dentofacial to isolated dental malocclusions.
The sheer multiplicity of genes involved in craniofacial and specifically dental development encompasses a large range of variation responsible for changes in individual appearance, function, susceptibility to diseases, and response to treatment. This review explores how such genetic variation resulting from single nucleotide polymorphisms (SNPs), mutations in single or multiple genes as well as altered gene regulation can lead to vastly different phenotypic expression of the face and dentition.
Genetic Disorders Specific to Teeth
Tooth development represents a distinctive example of organogenesis, orchestrated by multiple cell types that progress through a series of morphological and functional stages. Central to this process is a carefully coordinated, sequential series of reciprocal epithelial -mesenchyme interactions which are facilitated by an intricate communication between different genetic pathways and signaling molecules [14]. Explicably, deficiencies in one or more of these processes can lead to a wide range of dental abnormalities affecting the number of teeth, size/shape of teeth, structural abnormalities of individual mineralized tissues as well as eruption abnormalities.
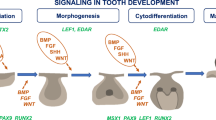
While numerous environmental influences can result in dental abnormalities (prenatal/postnatal nutrition, chemotherapy, radiotherapy, infections, trauma, pathologies etc.), genetic and epigenetic factors play a major role, making the developmental and disease process truly multifactorial [15]. Collectively, more than a few hundred genes have been recognized as associated with tooth development [16, 17]. Among these, the WNT, BMP, FGF, and HH families of receptors, ligands, and intracellular signaling effectors are recurrently utilized and are presently considered the key signaling pathways in odontogenesis. Expression patterns of these genes, along the different tooth developmental stages, are available in a database created at the University of Helsinki (http://bite-it.helsinki.fi). The resultant genetic spectrum of any disorder is defined by a large range of normal variation that leads to changes in individual appearance, function, susceptibility to diseases, and response to treatments (Fig. 1).

Dentofacial phenotypes can be found within a spectrum of severity: normal variation, isolated dental abnormities, co-segregation with other non-dental abnormalities, or as part of a more severe syndrome. Genetic, epigenetic, and environmental factors contribute individually or jointly to this spectrum
Dental abnormalities can also appear in a fairly isolated manner and are usually diagnosed and treated by a dental practitioner. Briefly, isolated abnormalities can be familial (inherited in a recessive, dominant, x-linked or multifactorial manner) or sporadic—caused by stochastic, de novo mutations in an individual. Alternatively, dental abnormalities frequently present as part of larger genetic disorder or syndrome. This fact is not surprising because the highly conserved signaling pathways of tooth development are also involved in the morphogenesis and patterning of other tissues, organs, and structures [16, 18].
Isolated Dental Disorders and/or Those Co-Segregating with a Larger Syndrome
Recent studies have revealed that mutations in the same genes can lead to both, isolated dental disorders and those which co-segregate with a syndrome. In such instances, it is not only the gene identity, but also the specific type and/or localization of the mutation, which dictates the severity of a disease and spectrum of the phenotype. A good example of this occurrence is the genetic determinants of tooth agenesis. In humans, tooth agenesis refers to the congenital absence of teeth (including hypodontia, <6 missing teeth, oligodontia >6 missing teeth or anodontia, complete absence of teeth) with hypodontia being observed most frequently [19]. Notably, some form of tooth agenesis appears as a phenotype in more than 150 syndromes [20].
The MSX1 gene (muscle segment homeobox 1 which encodes for MSX1) was one of the first genes to be linked with tooth agenesis [21]. Molecular genetic analyses over the years have elaborated on the genotype-phenotype correlation between MSX1 mutations and tooth agenesis [22]. For instance, a recent study found a novel non-stop MSX1 mutation in a Chinese family [23], which results in an inappropriately elongated MSX1 protein. Another nonsense mutation Trp139Stop was discovered in a Japanese family with hypodontia/oligodontia [24]. In all instances, there is a selective bilateral loss of posterior teeth, mainly the second premolar and third molars. Moreover, the dental phenotype appears fairly isolated in most affected individuals. A more severe phenotype has been reported however, when a nonsense mutation—Ser202Stop—results in a truncated MSX1 protein lacking the C-terminal end of the homeodomain, causing dysplastic tooth and nail formation (part of Witkop syndrome) [25] and when there is a complete loss of the MSX1 homeodomain resulting in tooth agenesis and orofacial clefts [26]. One scenario we report here is illustrated by the segregation of an MSX1frameshift mutation (Glu78FS) in a nuclear family resulting in a more severe tooth agenesis phenotype than a missense mutation at the same residue (Fig. 2a–d). A similar genotype-phenotype correlation is found for PAX9 mutations wherein nonsense and frameshift mutations give rise to a more severe phenotype than missense mutations [27, 28]. These correlations are not only useful for diagnostic purposes but also provide fundamental information into the mechanisms governing the functions of individual gene products in tooth development.


a Representative chromatogram from an individual with severe tooth agenesis. Comparison of the WT MSX1 sequence (above) and the mutated DNA sequence (below) of the MSX1 gene that was found to contain a complex rearrangement, GAG deletion, and C insertion at nucleotide 250 that resulted in a frameshift mutation and a premature stop codon. b The mutation described above co-segregates in an autosomal dominant manner with a severe form of tooth agenesis in a nuclear family as shown in the pedigree (shaded symbols = affected, circle = female, squares = male, and proband indicated with an arrow). c Panoramic image shows a representative phenotypic pattern seen in the proband involving premolars and molars. d Multi-alignment of the MSX1 protein (amino acid sequence) showing the Glu78FS mutation segregating with tooth agenesis in one family as compared to multiple species (human, mouse and cow MSX1 and MSX2). Other mutations previously reported in tooth agenesis and CL/P are indicated and labeled above the Glu78FS sequence. The observation that our frameshift mutation at Glu78FS resulted in a more severe tooth agenesis phenotype than a missense mutation at the same residue (Glu78Val) would be expected since the frameshift mutation resulted in a missing homeodomain region and likely led to the disruption of folding of most or the entire protein. Sequence alignment of Glu78FS with other reported mutations in MSX1 suggests that mutations leading to tooth agenesis result when the function of the homeodomain is destroyed, either directly due to a mutation within the homeodomain itself (Arg196Pro, Ser202Stop), or indirectly as a result of a stop mutation (Ser104X). We can further speculate from this observation that the M61K mutation, which is a point mutation not directly affecting the homeodomain, may define a region which interacts with the homeodomain. Conversely, all of the point mutations that lead to oral clefting shown in green highlight are found in the region including residues 78 through 151. We speculate the latter may define a region important in the formation of oral clefting
Similar to tooth agenesis, supernumerary teeth can occur either as part of a larger disease or syndrome or they can present as an idiopathic finding. [29], Lubinsky et al. [30••] reviewed the existing literature to find evidence of specific associations between supernumerary teeth and syndromes and reported that eight syndromes have strong evidence for an association: cleidocranial dysplasia, familial adenomatous polyposis, trichorhinophalangeal syndrome, type I; Rubinstein–Taybi syndrome, Nance–Horan syndrome, Opitz BBB/G syndrome, oculofaciocardiodental syndrome, and autosomal dominant Robinow syndrome. A related but perhaps independent occurrence is seen in patients with cleft lip and palate (CLP) who present with associated dental abnormalities including supernumerary teeth, hypodontia, enamel hypoplasia, taurodontism, and altered crown shape and size [31]. The putative association of supernumerary teeth and orofacial clefts may be a non-specific consequence of the bony disruptions [32], but Chu et al. [33] found abnormal crown and root patterning, taurodontism, variable hypodontia and supernumerary teeth as well as reduced enamel density after conditionally knocking-out Irf6. Although this does not rule out the explanation of an indirect environmental consequence of tooth number defects due to the proximity of orofacial clefts, it does attribute a more direct effect of a genetic defect (in this case IRF6) to the observed dental anomalies.
As discussed above, the same genes that result in a dental disorder co-segregating with a syndrome can also cause isolated dental disorders. For example, mutations in WNT10A were identified in 25–50% of isolated hypodontia cases depending on the specific population tested [34, 35]. Unlike MSX1 and PAX9, premolars were the most frequently missing teeth in individuals with WNT10A mutations. Moreover, biallelic WNT10A mutations were associated with missing molars as well as mandibular central incisors [34]. Indeed, the genotype-phenotype correlation of tooth agenesis has been studied more than most isolated dental disorders. But, examples of isolated dental disorders are not limited to tooth agenesis. For instance, disorders of tooth eruption represent the archetypal scenario where the genetic basis of the eruption failure was overlooked for other explanations of causality, such as ankylosis or mechanical obstruction [36, 37]. More specifically, primary failure of eruption (PFE-OMIM 125350), was previously thought to be sporadic and not inherited, but in fact is due to a genetic mutation in the PTH1R gene [36, 38].
The intrigue of the PFE story lies not in the fact that this isolated dental condition is caused by genetic mutations in PTH1R, but that similar to the examples described in tooth agenesis above, one gene, PTH1R, when mutated can cause either a lethal syndrome or a dental-specific eruption disorder (discussed below). The etiology of eruption disorders includes local and non-syndromic causes (cysts, ankylosis, lateral tongue pressure, supernumerary teeth, thumb habit) [39,40,41,42] but also by co-segregation with a genetic syndrome such as cleidocranial dysplasia, Hunter’s disease, and osteopetrosis (reviewed by Wise et al. [43]).
Despite the strides made to definitively characterize PFE and other eruption disorders, a clear understanding remains elusive. First described by Proffit and Vig, canonical reports describe PFE as a local/non-syndromic eruption disorder marked by a failure of permanent (adult) tooth eruption in the absence of mechanical obstruction affecting only the posterior dentition [39]. The use of the term “primary” originated from the clinical observation that the defect is presumed to be within the eruption mechanism of the tooth and bony crypt, since there is no apparent mechanical interference.
Discovery of at least one gene that causes PFE and the clear segregation of the PFE phenotype within families established the genetic basis of PFE by mutations in PTH1R [36, 38]. Nonetheless, the specific molecular mechanisms that orchestrate a normal eruption process are poorly understood. And, the pathogenesis of PTH1R mutations in PFE is unknown. For example, there is no clear explanation as to why PFE, caused by genetic mutations in PTH1R, may result in either uni- or bilateral eruption failure, or include involvement of the maxilla or mandible. Furthermore, mutations in the PTH1R gene present in clinically diverse conditions, such as Jansen’s metaphyseal chondrodysplasia (OMIM ID: 156400), Blomstrand’s lethal chondrodysplasia (OMIM ID: 215045), enchondromatosis (Ollier’s Disease) (OMIM ID: 166,000), osteoarthritis, and PFE represent a breakthrough in the dental and orthodontic communities.
Subsequently, the knowledge that PFE occurs as an isolated condition due to a mutation in the same gene that causes a lethal syndrome creates a paradigm shift in how we diagnose and therefore manage dental anomalies. Specifically, this alerts the clinician that the adverse response of PFE-affected teeth to orthodontic treatment would be the intrusion of adjacent teeth and no movement of affected teeth. The distinction of a genetically based PFE diagnosis is critical; if a tooth is instead misdiagnosed as “ankylosed,” or fused to the bone based on clinical parameters, it will lead to inappropriate treatment and the chance for success is futile. Hence, our advances in genotype-phenotype studies have also cautioned us to approach clinical scenarios of eruption disorders with a more discerning diagnostic rubric and therefore appropriate treatment plan.
The genetic basis of tooth eruption also presents a unique opportunity for investigating the fundamental mechanisms that dictates tooth-bone interactions. For instance, the PTH1R gene is critical in the skeletal and endocrine systems, but there is a paucity of information explaining the role in the dental tissues. We already know that PTHrP has been shown to be integral in the process of tooth eruption based on studies in the mouse model; the presence of PTHrP was shown in the enamel organ and especially the stellate reticulum of the dental follicle [44]. Furthermore, studies documenting the conditional-PTHrP knockout mouse revealed that otherwise normal developing teeth became impacted and encapsulated by a bony crypt. Central to this process in tooth eruption is the activation of the cAMP/PKA pathway (via Gs protein activation) by either ligand resulting in the progression of tooth development and eruption—while the disruption of the same pathway results in a cessation of eruption (upregulation of the biomineralization of cementoblasts) [45].
The PFE/PTH1R story does not end with the initial genotype-phenotype accomplishments described above. The apparent crosstalk between eruption and other tooth developmental process was described by Ono et al. (2016) who investigated dental root formation using a lineage-tracing model [46]. Ono and collaborators found that cells in the dental follicle and root surface express parathyroid hormone-related peptide (PTHrP), and deletion of the PTHrP receptor (PPR) in these progenitors leads to failure of eruption and significantly truncated roots lacking periodontal ligaments. The juxtaposition of eruption failure and root formation sparks the question of whether there is a parallel phenomena in humans. Remarkably, reports of short root anomaly in humans failed to show the co-segregation of shortened roots and eruption failure, but categorically provides an interesting model to explore what, if any, epistatic interactions lead to one condition (i.e., short roots) in the absence of the other (i.e., eruption failure) in humans versus mice [47].
Epigenetic Regulation in Dental Development
Epigenetic analyses provide viable alternative theories to explain apparent genetic etiology in dentofacial development. For example, the epigenetic architecture of patients with X-linked hypohidrotic ectodermal dysplasia (XLHED) present with an unclear correlation between the severity of mutations in ectodysplasin A (EDA) gene and the observed severity of the resultant XLHED phenotype. XLHED patients have conspicuous clinical features: small, oddly shaped or missing teeth, decreased salivary flow, reduced sweating, and sparse hair and often carry distinctive craniofacial features. Distinct mutations in the EDA gene [48, 49] have been cited as responsible for XLHED, but the correlation is unlike the results of specific MSX1 mutations described above. In fact, moderate, minor or an absence of features has been reported in individuals who are heterozygous carriers of an EDA mutation [50]. Yin et al. found an epigenetic control of EDA (via cytosine methylation) was due to large CpG islands within the promoter of EDA; its transcription can be significantly modulated depending by the degree of methylation. [51]. The CpG rich EDA promoter region was found to be hypermethylated in ~78% of carriers of XLHED [51], a modification which could presumably inhibit expression of the mutated gene product. A similar finding was reported when investigators completed a DNA methylation profiling of patients with non-syndromic tooth agenesis [52]. They found a group of differentially methylated promoters in novel genes in affected patients compared to controls. While the obvious, hypodontia-related genes (MSX, PAX9, EDA, AXIN) did not display significant methylation status differences, some of the identified genes do participate in signaling pathways important for tooth development [53••].
Another level of epigenetic control can be found in gene regulation via microRNAs (miRNAs), and it is thought to influence the expression of almost 30% of all protein-coding genes [54]. miRNAs have been found to be differentially expressed in dental epithelial and mesenchyme tissues of different tooth types [55,56,57,58] and participate in the differentiation of odontogenic cells including ameloblasts, odontoblasts, dental follicular, and pulp cells [55, 59,60,61, 58]. Abnormal dental phenotypes have also been found in mouse studies where microRNA function is disrupted by knockout of Dicer1, which is required for miRNA maturation. Using Pitx2-Cre mice, Dicer1 was conditionally knocked-out in dental epithelial cells at an early stage [55]. This resulted in the upregulation of several hundred genes, with increased proliferation and decreased differentiation of progenitor cells. Moreover, knockout animals had prominent dental anomalies: extra incisors, severely misshapen teeth, enamel-free incisors, cuspless molars as well as a uniquely “branched” incisor [55]. Recently, Fan et al. [62] injected a soluble miR-224 agomir (miRNA mimics) into incisors of neonatal mice and found significant structural differences in enamel, including disorganization of enamel prism structure, deficient crystal growth, and reduced microhardness. Studies such as these highlight the role of microRNAs in tooth development and suggest that disturbances in microRNAs may result in tooth developmental abnormalities.
Current and Future Milestones
Molecular Diagnosis and Pharmacological Interventions to Manage Dental Disorders
We anticipate that dentistry, similar to medicine, will transform clinical practice based on genetic knowledge. To this end, Prasad et al. [62] are utilizing contemporary next-generation sequencing (NGS) methods to develop array panels that specifically target isolated and syndrome-associated dental disorders. Their panel consists of genes compiled from human and animal studies that are known to be involved in tooth development as well as diseases with orodental phenotypes [63••]. In preliminary studies using a pilot panel in a cohort of 101 patients with no molecular diagnosis of orodental abnormalities, mutations were identified in 39 of the cohort with several novel pathogenic variants [63••]. As our knowledge of genetic and epigenetic control of tooth developmental disorders improves, such panels can be enlarged and employed for diagnostic purposes.
Taken together, advances in genetics, molecular and pharmacological research, coupled with the utilization of animal models, are indeed paving the way for novel interventions to manage human genetic developmental disorders. A great example is the multitude of research into abovementioned XHLED, and its causative mutations in the ectodysplasin A gene (EDA). When tested in a dog model of XLHED, postnatal intravenous administration of soluble recombinant EDA (to replace the mutant EDA-A1) was tolerated well and provided a significant improvement in phenotype, including the stable restoration of permanent dentition and recovered function of secretory glands [64]. The FDA has now approved the recombinant protein (EDI200) for human studies [65], which promises to be a therapeutic intervention for patients suffering from XLHED.
Conclusion
It is clear from the examples stated above that many genetic diseases involving the dental tissues have varied clinical presentations. Indeed, a dental finding may be the singular early discovery that leads to the diagnosis of a more complex gene defect. However, it is critical to make a distinction between unrelated dental findings observed with other phenotypes, as highlighted by Lubinsky et al. [30••].
In this post-genomic era, the role of epigenetic regulatory mechanisms in controlling various developmental processes has an important impact on human dentofacial development. Recent clinical studies utilizing large cohorts of twins are proving valuable in looking beyond genetics, highlighting the influences of epigenetics in phenotype determination [66,67,68]. Moreover, declining costs of genome sequencing allows increasing opportunities to analyze the unique genotype-phenotype relationships for the purposes of diagnosis, prevention, and treatment of dentofacial abnormalities. A potential bottleneck in appropriately utilizing this genetic information stems from our dissimilar and often ambiguous methods to identify, categorize, and describe dentofacial anomalies and phenotypes. Certainly, as the above-described NGS-based methods for identifying genetic mutations [63••] become more refined and widely utilized, precise phenotyping will be paramount for the accuracy and efficacy of these novel diagnostic tools. Future efforts should be directed toward more unambiguous methods of reporting and defining findings.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Gans C, Northcutt RG. Neural crest and the origin of vertebrates: a new head. Science. 1983;220(4594):268–73. doi:10.1126/science.220.4594.268.
Szabo-Rogers HL, Smithers LE, Yakob W, Liu KJ. New directions in craniofacial morphogenesis. Dev Biol. 2010;341(1):84–94. doi:10.1016/j.ydbio.2009.11.021.
Trainor PA, Richtsmeier JT. Facing up to the challenges of advancing craniofacial research. Am J Med Genet A. 2015;167(7):1451–4. doi:10.1002/ajmg.a.37065.
Carlson DS. Evolving concepts of heredity and genetics in orthodontics. Am J Orthod Dentofac Orthop. 2015;148(6):922–38. doi:10.1016/j.ajodo.2015.09.012.
Harris JE, Kowalski CJ. All in the family: use of familial information in orthodontic diagnosis, case assessment, and treatment planning. Am J Orthod. 1976;69(5):493–510.
Hunter WS, Balbach DR, Lamphiear DE. The heritability of attained growth in the human face. Am J Orthod. 1970;58(2):128–34.
Watson WG. Hereditary environment. Am J Orthod. 1980;78(3):331–3.
Feldman MW, Lewontin RC. The heritability hang-up. Science. 1975;190(4220):1163–8.
Corruccini RS, Potter RH. Genetic analysis of occlusal variation in twins. Am J Orthod. 1980;78(2):140–54.
Harris EF, Johnson MG. Heritability of craniometric and occlusal variables: a longitudinal sib analysis. Am J Orthod Dentofac Orthop. 1991;99(3):258–68. doi:10.1016/0889-5406(91)70007-J.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi:10.1038/35057062.
Ragoussis J. Genotyping technologies for genetic research. Annu Rev Genomics Hum Genet. 2009;10:117–33. doi:10.1146/annurev-genom-082908-150116.
Giannobile WV, Kornman KS, Williams RC. Personalized medicine enters dentistry: what might this mean for clinical practice? J Am Dent Assoc. 2013;144(8):874–6.
Balic A, Thesleff I. Tissue interactions regulating tooth development and renewal. Curr Top Dev Biol. 2015;115:157–86. doi:10.1016/bs.ctdb.2015.07.006.
Brook AH. Multilevel complex interactions between genetic, epigenetic and environmental factors in the aetiology of anomalies of dental development. Arch Oral Biol. 2009;54(Suppl 1):S3–17. doi:10.1016/j.archoralbio.2009.09.005.
Bei M. Molecular genetics of tooth development. Current opinion in genetics & development. 2009;19(5):504–10. doi:10.1016/j.gde.2009.09.002.
Thesleff I. The genetic basis of tooth development and dental defects. Am J Med Genet A. 2006;140(23):2530–5. doi:10.1002/ajmg.a.31360.
Lan Y, Jia S, Jiang R. Molecular patterning of the mammalian dentition. Semin Cell Dev Biol. 2014;25-26:61–70. doi:10.1016/j.semcdb.2013.12.003.
Polder BJ, Van't Hof MA, Van der Linden FP, Kuijpers-Jagtman AM. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dent Oral Epidemiol. 2004;32(3):217–26. doi:10.1111/j.1600-0528.2004.00158.x.
Yin W, Bian Z. The gene network underlying hypodontia. J Dent Res. 2015;94(7):878–85. doi:10.1177/0022034515583999.
Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet. 1996;13(4):417–21. doi:10.1038/ng0896-417.
Mostowska A, Kobielak A, Trzeciak WH. Molecular basis of non-syndromic tooth agenesis: mutations of MSX1 and PAX9 reflect their role in patterning human dentition. Eur J Oral Sci. 2003;111(5):365–70.
Wong SW, Liu HC, Han D, Chang HG, Zhao HS, Wang YX, et al. A novel non-stop mutation in MSX1 causing autosomal dominant non-syndromic oligodontia. Mutagenesis. 2014;29(5):319–23. doi:10.1093/mutage/geu019.
Kimura M, Machida J, Yamaguchi S, Shibata A, Tatematsu T, Miyachi H, et al. Novel nonsense mutation in MSX1 in familial nonsyndromic oligodontia: subcellular localization and role of homeodomain/MH4. Eur J Oral Sci. 2014;122(1):15–20. doi:10.1111/eos.12105.
Jumlongras D, Bei M, Stimson JM, Wang WF, DePalma SR, Seidman CE, et al. A nonsense mutation in MSX1 causes Witkop syndrome. Am J Hum Genet. 2001;69(1):67–74. doi:10.1086/321271.
van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet. 2000;24(4):342–3. doi:10.1038/74155.
Frazier-Bowers SA, Guo DC, Cavender A, Xue L, Evans B, King T, et al. A novel mutation in human PAX9 causes molar oligodontia. J Dent Res. 2002;81(2):129–33.
Bailleul-Forestier I, Molla M, Verloes A, Berdal A. The genetic basis of inherited anomalies of the teeth. Part 1: clinical and molecular aspects of non-syndromic dental disorders. European journal of medical genetics. 2008;51(4):273–91. doi:10.1016/j.ejmg.2008.02.009.
Anthonappa RP, King NM, Rabie AB. Prevalence of supernumerary teeth based on panoramic radiographs revisited. Pediatr Dent. 2013;35(3):257–61.
•• Lubinsky M, Kantaputra PN. Syndromes with supernumerary teeth. Am J Med Genet A. 2016; doi:10.1002/ajmg.a.37763. Analyzes the evidence of assoications between genetic syndromes and supernumerary teeth
Subasioglu A, Savas S, Kucukyilmaz E, Kesim S, Yagci A, Dundar M. Genetic background of supernumerary teeth. European journal of dentistry. 2015;9(1):153–8. doi:10.4103/1305-7456.149670.
Garvey MT, Barry HJ, Blake M. Supernumerary teeth—an overview of classification, diagnosis and management. Journal. 1999;65(11):612–6.
Chu EY, Tamasas B, Fong H, Foster BL, LaCourse MR, Tran AB, et al. Full spectrum of postnatal tooth phenotypes in a novel Irf6 cleft lip model. J Dent Res. 2016; doi:10.1177/0022034516656787.
Arzoo PS, Klar J, Bergendal B, Norderyd J, Dahl N. WNT10A mutations account for (1/4) of population-based isolated oligodontia and show phenotypic correlations. Am J Med Genet A. 2014;164A(2):353–9. doi:10.1002/ajmg.a.36243.
van den Boogaard MJ, Creton M, Bronkhorst Y, van der Hout A, Hennekam E, Lindhout D, et al. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet. 2012;49(5):327–31. doi:10.1136/jmedgenet-2012-100750.
Frazier-Bowers SA, Simmons D, Wright JT, Proffit WR, Ackerman JL. Primary failure of eruption and PTH1R: the importance of a genetic diagnosis for orthodontic treatment planning. Am J Orthod Dentofac Orthop. 2010;137(2):160 e1-7. doi:10.1016/j.ajodo.2009.10.019.discussion -1
Frazier-Bowers SA, Puranik CP, Mahaney MC. The etiology of eruption disorders—further evidence of a ‘genetic paradigm’. Semin Orthod. 2010;16(3):180–5. doi:10.1053/j.sodo.2010.05.003.
Decker E, Stellzig-Eisenhauer A, Fiebig BS, Rau C, Kress W, Saar K, et al. PTHR1 loss-of-function mutations in familial, nonsyndromic primary failure of tooth eruption. Am J Hum Genet. 2008;83(6):781–6. doi:10.1016/j.ajhg.2008.11.006.
Proffit WR, Vig KW. Primary failure of eruption: a possible cause of posterior open-bite. Am J Orthod. 1981;80(2):173–90.
Rhoads SG, Hendricks HM, Frazier-Bowers SA. Establishing the diagnostic criteria for eruption disorders based on genetic and clinical data. Am J Orthod Dentofac Orthop. 2013;144(2):194–202. doi:10.1016/j.ajodo.2013.03.015.
S Frazier-Bowers. Primary failure of eruption: clinical implications of a genetic disorder. Effective and efficient tooth movement: evidence based orthodontics 48th ed Ann Arbor, MI:. 2011;McNamara JA Jr, Hatch N, Kapila SD, editors(Craniofacial Growth Series, Department of Orthodontics and Pediatric Dentistry and Center for Human Growth and Development. 2011).
Juppner P. Pediatric bone: biology & diseases: Gulf Professional Publishing; 2003.
Wise GE, Frazier-Bowers S, D'Souza RN. Cellular, molecular, and genetic determinants of tooth eruption. Crit Rev Oral Biol Med. 2002;13(4):323–34.
Fukushima H, Jimi E, Kajiya H, Motokawa W, Okabe K. Parathyroid-hormone-related protein induces expression of receptor activator of NF-{kappa}B ligand in human periodontal ligament cells via a cAMP/protein kinase A-independent pathway. J Dent Res. 2005;84(4):329–34.
Ouyang H, McCauley LK, Berry JE, Saygin NE, Tokiyasu Y, Somerman MJ. Parathyroid hormone-related protein regulates extracellular matrix gene expression in cementoblasts and inhibits cementoblast-mediated mineralization in vitro. J Bone Miner Res. 2000;15(11):2140–53. doi:10.1359/jbmr.2000.15.11.2140.
Ono W, Sakagami N, Nishimori S, Ono N, Kronenberg HM. Parathyroid hormone receptor signalling in osterix-expressing mesenchymal progenitors is essential for tooth root formation. Nat Commun. 2016;7:11277. doi:10.1038/ncomms11277.
Puranik CP, Hill A, Henderson Jeffries K, Harrell SN, Taylor RW, Frazier-Bowers SA. Characterization of short root anomaly in a Mexican cohort—hereditary idiopathic root malformation. Orthod Craniofac Res. 2015;18(Suppl 1):62–70. doi:10.1111/ocr.12073.
Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, Ferguson B, et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet. 1996;13(4):409–16. doi:10.1038/ng0895-409.
Schneider P, Street SL, Gaide O, Hertig S, Tardivel A, Tschopp J, et al. Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J Biol Chem. 2001;276(22):18819–27. doi:10.1074/jbc.M101280200.
Vincent MC, Biancalana V, Ginisty D, Mandel JL, Calvas P. Mutational spectrum of the ED1 gene in X-linked hypohidrotic ectodermal dysplasia. European journal of human genetics : EJHG. 2001;9(5):355–63. doi:10.1038/sj.ejhg.5200635.
Yin W, Ye X, Fan H, Bian Z. Methylation state of the EDA gene promoter in Chinese X-linked hypohidrotic ectodermal dysplasia carriers. PLoS One. 2013;8(4):e62203. doi:10.1371/journal.pone.0062203.
Palmke N, Santacruz D, Walter J. Comprehensive analysis of DNA-methylation in mammalian tissues using MeDIP-chip. Methods. 2011;53(2):175–84. doi:10.1016/j.ymeth.2010.07.006.
•• Wang J, Sun K, Shen Y, Xu Y, Xie J, Huang R, et al. DNA methylation is critical for tooth agenesis: implications for sporadic non-syndromic anodontia and hypodontia. Scientific reports. 2016;6:19162. doi:10.1038/srep19162. Pilot study evaluating genomic DNA methylation profiles in patients with hypodontia
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi:10.1016/j.cell.2004.12.035.
Cao H, Wang J, Li X, Florez S, Huang Z, Venugopalan SR, et al. MicroRNAs play a critical role in tooth development. J Dent Res. 2010;89(8):779–84. doi:10.1177/0022034510369304.
Michon F, Tummers M, Kyyronen M, Frilander MJ, Thesleff I. Tooth morphogenesis and ameloblast differentiation are regulated by micro-RNAs. Dev Biol. 2010;340(2):355–68. doi:10.1016/j.ydbio.2010.01.019.
Jevnaker AM, Osmundsen H. MicroRNA expression profiling of the developing murine molar tooth germ and the developing murine submandibular salivary gland. Arch Oral Biol. 2008;53(7):629–45. doi:10.1016/j.archoralbio.2008.01.014.
Yin K, Hacia JG, Zhong Z, Paine ML. Genome-wide analysis of miRNA and mRNA transcriptomes during amelogenesis. BMC Genomics. 2014;15:998. doi:10.1186/1471-2164-15-998.
Liu H, Lin H, Zhang L, Sun Q, Yuan G, Zhang L, et al. miR-145 and miR-143 regulate odontoblast differentiation through targeting Klf4 and Osx genes in a feedback loop. J Biol Chem. 2013;288(13):9261–71. doi:10.1074/jbc.M112.433730.
Khan QE, Sehic A, Khuu C, Risnes S, Osmundsen H. Expression of Clu and Tgfb1 during murine tooth development: effects of in-vivo transfection with anti-miR-214. Eur J Oral Sci. 2013;121(4):303–12. doi:10.1111/eos.12056.
Chen P, Wei D, Xie B, Ni J, Xuan D, Zhang J. Effect and possible mechanism of network between microRNAs and RUNX2 gene on human dental follicle cells. J Cell Biochem. 2014;115(2):340–8. doi:10.1002/jcb.24668.
Fan Y, Zhou Y, Zhou X, Sun F, Gao B, Wan M, et al. MicroRNA 224 regulates ion transporter expression in ameloblasts to coordinate enamel mineralization. Mol Cell Biol. 2015;35(16):2875–90. doi:10.1128/MCB.01266-14.
•• Prasad MK, Geoffroy V, Vicaire S, Jost B, Dumas M, Le Gras S, et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J Med Genet. 2016;53(2):98–110. doi:10.1136/jmedgenet-2015-103302. Targeted next-generation sequencing assay to identify genes mutated in orodental disorders
Casal ML, Lewis JR, Mauldin EA, Tardivel A, Ingold K, Favre M, et al. Significant correction of disease after postnatal administration of recombinant ectodysplasin A in canine X-linked ectodermal dysplasia. Am J Hum Genet. 2007;81(5):1050–6. doi:10.1086/521988.
Huttner K. Future developments in XLHED treatment approaches. Am J Med Genet A. 2014;164A(10):2433–6. doi:10.1002/ajmg.a.36499.
Hughes TE, Townsend GC, Pinkerton SK, Bockmann MR, Seow WK, Brook AH, et al. The teeth and faces of twins: providing insights into dentofacial development and oral health for practising oral health professionals. Aust Dent J. 2014;59(Suppl 1):101–16. doi:10.1111/adj.12101.
Townsend G, Bockmann M, Hughes T, Brook A. Genetic, environmental and epigenetic influences on variation in human tooth number, size and shape. Odontology/the Society of the Nippon Dental University. 2012;100(1):1–9. doi:10.1007/s10266-011-0052-z.
Hughes T, Bockmann M, Mihailidis S, Bennett C, Harris A, Seow WK, et al. Genetic, epigenetic, and environmental influences on dentofacial structures and oral health: ongoing studies of Australian twins and their families. Twin research and human genetics: the official journal of the International Society for Twin Studies. 2013;16(1):43–51. doi:10.1017/thg.2012.78.
Acknowledgements
S.R.V is supported by the Post-doctoral Fellowship Award from the American Association of Orthodontists Foundation. We gratefully acknowledge the participation of the family in the MSX1 tooth agenesis study (SAFB), the technical assistance of Drs. Dongchuan Guo and Brenda Temple and the support of NIH 1K23RR17442.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Sylvia Frazier-Bowers declares no conflict of interest.
Siddharth Vora reports grants from American Association of Orthodontists Foundation (AAOF).
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Craniofacial Skeleton
Rights and permissions
About this article

Cite this article
Frazier-Bowers, S.A., Vora, S.R. Genetic Disorders of Dental Development: Tales from the Bony Crypt. Curr Osteoporos Rep 15, 9–17 (2017). https://doi.org/10.1007/s11914-017-0342-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-017-0342-7