
Abstract
Significant advances have been made in the management of pulmonary arterial hypertension (PAH) in the past decade. There is a greater understanding of the disease process, more robust markers of prognostication and a wider range of disease-targeted therapies, with three classes of drug therapy now established. This has resulted in improved prognosis and quality of life but has also increased the complexity in making treatment decisions. To utilise these pharmacotherapies to their best potential, several factors need to be considered. This article will discuss how best to initiate and escalate PAH therapy on an individual patient basis by discussing current pharmacotherapies available, specific patient factors and determining treatment response and prognosis at diagnosis and during follow-up.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pulmonary arterial hypertension (PAH) is a progressive, incurable vasculopathy of the pulmonary circulation. Pulmonary artery remodelling results in a progressive rise in pulmonary vascular resistance (PVR) and right ventricular (RV) afterload, leading to RV dilatation, RV failure and eventually premature death.
Pulmonary hypertension (PH) is defined by a mean pulmonary artery pressure (mPAP) ≥25 mmHg at rest, measured during right heart catheterisation. Patients with PH are classified into five groups based on underlying pathogenesis of the disease. PAH describes WHO group 1 disease and is additionally characterised hemodynamically by a pulmonary artery wedge pressure (PAWP) ≤15 mmHg and PVR >3 Wood units as outlined in the Nice 2013 clinical classification of PH [1]:
-
1.
Pulmonary arterial hypertension
-
1.1
Idiopathic PAH
-
1.2
Heritable PAH
-
1.2.1
ALK-1
-
1.2.2
ENG, SMAD9, CAV1, KCNK3
-
1.2.3
Unknown
-
1.2.1
-
1.3
Drug and toxin induced
-
1.4
Associated with
-
1.4.1
Connective tissue disease (CTD-PAH)
-
1.4.2
HIV infection
-
1.4.3
Portal hypertension (PoP-PAH)
-
1.4.4
Congenital heart disease (CTD-PAH)
-
1.4.5
Schistosomiasis
-
1.4.1
-
1.1′
Pulmonary veno-occlusive disease and/or pulmonary capillary haemangiomatosis
-
1.1′′
Persistent PH of the newborn (PPHN)
-
1.1
-
2.
Pulmonary hypertension due to left heart disease
-
3.
Pulmonary hypertension due to lung disease and / or hypoxia
-
4.
Chronic thromboembolic pulmonary hypertension (CTEPH)
-
5.
Pulmonary hypertension with unclear multifactorial mechanisms
- BMPR:
-
bone morphogenic protein receptor type II
- CAV1:
-
caveolin-1
- ENG:
-
endoglin
- HIV:
-
human immunodeficiency virus
- PAH:
-
pulmonary arterial hypertension
Correct diagnosis is crucial, as the prognosis and management for each category is different [2]. The medical management and understanding of the pathobiology of PAH has evolved significantly over the past 20 years. With the advent of three distinct classes of effective drug therapy, there has been a consequent improvement in survival. Median survival was 2.8 years from diagnosis prior to modern disease-targeted treatment [3], prognosis has now improved, with a 3-year survival of 54.5 % [4], compared with 48 % in 1991.
There remains a significant morbidity and mortality associated with PAH that requires further intervention to improve survival. As well as ongoing research into developing new therapies, it is important that the current therapies are used optimally. To do this, patients require specialist care and accurate prognostication of their disease, with an individually tailored treatment strategy appropriate to their clinical condition.
This article will focus on how to determine the optimal treatment for a patient with pulmonary hypertension by addressing the following areas:
-
Current disease-targeted therapies available
-
Determining prognosis in PAH and assessing response to treatment
-
Treatment goals and treatment algorithm in PAH
-
The role of combination therapy
-
Future areas of development in PAH therapy
Disease-Targeted Therapies in Pulmonary Arterial Hypertension
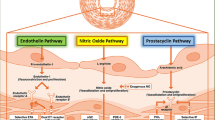
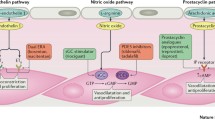
Three main pathobiological pathways are targeted with current disease-specific PAH therapy: the prostacyclin pathway, the nitric oxide pathway and the endothelin pathway (Fig. 1). Other pathways such as the platelet-derived growth factor pathway are subject to ongoing investigation.

Pathobiological pathways in PAH. Reprinted from Humbert M et al. Treatment of pulmonary arterial hypertension. N Engl J Med, 351:1425–36. Copyright © 2004 with permission from Massachusetts Medical Society. Licence Number: 3584751356819
The currently available disease-targeted therapies for PAH were recently extensively reviewed by Wolfson et al. [5] and will be briefly reviewed here.
Prostanoids
Prostanoids can be administered intravenously (IV), subcutaneously (SC), by inhalation or orally. Due to their anti-platelet effect, the concurrent use of anti-platelets or anticoagulants may increase the risk of bleeding. Common side effects of parenteral prostanoids are headache, jaw pain, flushing, nausea, diarrhoea and musculoskeletal pain.
Epoprostenol
Epoprostenol is indicated for patients with symptomatic PAH (FC II-III) who have failed to respond to initial monotherapy or first line in patients in FC IV [6]. It is given by continuous IV infusion via a tunnelled central venous catheter. Epoprostenol has a short half-life; therefore, interruption of the infusion can cause potentially life-threatening rebound pulmonary hypertension. Chronic overdosing can result in high output cardiac failure. Aerosolized epoprostenol has been used in critically ill patients with pulmonary hypertension; however, there is not a strong enough evidence base to support its use in this context at present [7].
Treprostinil
Treprostinil, an analog of prostacyclin, has three licensed modes of delivery: SC, IV and inhaled, with oral therapy approved in the USA but not Europe. Advantageous properties of parenteral treprostinil are a longer half-life, thermostability and prefilled syringes exist that can be changed every 48 h. Transition from epoprostenol to treprostinil is both safe and effective [8], with roughly two times the dose of treprostinil vs epoprostinil being required. There is a reportedly higher incidence of catheter-associated bacteraemia in continuous IV treprostinil than epoprostenol [9]. Infusion site pain can limit SC therapy; however, more rapid titration reduces the incidence of this.
To tackle these problems, an implantable pump system (Lenus Pro®) has been developed to deliver continuous IV treprostinil. A pump is inserted in the abdominal wall fascia and a tunnelled catheter placed from this to the cephalic or subclavian vein. Cassettes are refilled using a gripper needle at 28-day intervals. Pilot data suggest this is a safe and effective method of delivering therapy, however requires a general anaesthetic for pump insertion [10].
Oral treprostinil has no clearly defined role in the PAH algorithm based on current evidence [11]. Inhaled treprostinil is licensed for WHO FC class III PAH. It has the advantage of four times daily dosing compared with the six to eight times daily dosing of iloprost.
Iloprost
Nebulised iloprost promotes selective pulmonary vasodilation, whilst minimising systemic side effects. Iloprost provides initial symptomatic improvement; however, only a minority of patients achieve long-term stability, particularly if it is used as monotherapy [12].
Beraprost
Beraprost is only available in Japan and South Korea. One small RCT has been conducted showing improved exercise capacity [13], with no haemodynamic benefit.
Selexipag
Selexipag is a novel orally active, non-prostanoid selective prostacyclin receptor agonist [14]. Data from the event driven RCT GRIPHON are awaited, pilot data suggest improvement in haemodynamics and exercise capacity.
Endothelin Receptor Antagonists
Endothelin receptor antagonists (ERA) are orally active drugs that bind to endothelin-A and -B receptors in pulmonary vascular smooth muscle cells. Three ERAs are currently approved for WHO FC II & III PAH [15, 16], Bosentan, Ambrisentan and Macitantan. Early therapy in patients with mildly symptomatic PAH has also been deemed efficacious [17]. Ambrisentan, a selective ET-A demonstrates a lower incidence of abnormal LFTs compared with Bosentan [18]. Macitentan, the newest ERA (2013), has sustained receptor binding and enhanced tissue penetration. No liver toxicity was seen in the RCT SERAPHIN [19••]; however, anaemia was more frequent. Sitaxsentan was withdrawn globally in 2011 due to cases of severe hepatotoxity [20].
ERAs are teratogenic; therefore, women of child-bearing age should use two forms of birth control therapy. There is an association with hepatocellular injury and anaemia; therefore, long-term use requires monthly monitoring of liver enzymes and regular full blood count. As a class effect, there is an association with peripheral oedema, headache, nasopharyngitis and gastrointestinal upset.
Nitric Oxide and Cyclic Guanosine Monophosphate Pathway
Impairment of nitric oxide (NO) synthesis and signaling through the NO-soluble guanylate cyclase (sGC)-guanosine monophosphate (cGMP) pathway is involved in the pathogenesis of PAH. Three main therapeutic targets exist:
-
1
Direct administration of inhaled NO
-
2
Increasing enzymatic production of NO: soluble guanylate cyclase (sGC) activators
-
3
Inhibition of NO metabolism: phosphodiesterase type 5 inhibitors (PDE5is)
Inhaled Nitric Oxide
Currently inhaled nitric oxide (iNO) has no role in the long-term management of PAH, having been assessed in pulsed nasal delivery and facemask form in small single-centre settings only [21, 22].
PDE5is
Inhibition of the cGMP degrading enzyme PDE5 results in vasodilation. Additionally, PDE5is exerts some anti-proliferative effects. Three drugs exist: Sildenafil, Tadalafil and Vardenafil. All improve exercise capacity, haemodynamics and symptoms but not mortality [23–25]. IV sildenafil is available for patients temporarily unable to ingest tablets. Transitioning to tadalafil from sildenafil due to adverse effects has been shown to be safe and well tolerated. Vardenafil, assessed only in the small RCT evaluation [26], is not yet approved for PAH.
PDE5is should be avoided in the context of recent stroke or myocardial infarction. Retinal and choroidal vasculatures express PDE5. There have been reports impaired of colour vision associated with sildenafil [27], therefore PDE5s should be avoided with a history of non-arteritic anterior ischaemic optic neuropathy or hereditary degenerative retinal disorders. Due to the risk of hypotension, the concomitant use of nitrates should be avoided. Most side effects are related to vasodilation, such as headache and flushing.
Soluble Guanylate Cyclase Stimulators
Riociguat is approved as monotherapy or in combination with ERAs for WHO FC II-III PAH. It has a dual mode of action, directly stimulating sGC independently of NO availability and acting in synergy with endogenous NO. Reported adverse events are haemoptysis and pulmonary haemorrhage [28], with common side effects being headache, dizziness and dyspepsia. Regular blood pressure monitoring is required during titration and the combination of riociguat and PDE5is is contraindicated due to the risk of hypotension.
Platelet-Derived Growth Factor Pathway
Proliferation of endothelial and vascular smooth muscle cells is a histopathological hallmark of PAH. Animal studies suggest that platelet-derived growth factor and c-KIT signaling are important in vascular smooth muscle cell proliferation and hyperplasia. Imatinib, an anti-proliferative tyrosine kinase inhibitor, improved exercise capacity and haemodynamics in two RCTS but with a high incidence of serious adverse events, such as subdural haemorrhage. As a consequence, imatinib is not licensed for use in pulmonary hypertension [29]
Treatment in Specific PAH Populations
CTD-PAH
Between 3 and 13 % of CTD patients will develop symptomatic PAH, with a higher incidence occurring in systemic sclerosis (SSc-PAH) [2]. CTD-PAH has a worse outcome compared with IPAH, with increased hospital admissions and reduced 1-year survival. Furthermore, the efficacy of disease-targeted therapy is reduced compared with other PAH cohorts [15] with the impact reduced most in SSc-PAH and those with associated interstitial lung disease [30]. There is some evidence that immunosuppression with cyclophosphamide and steroids result in haemodynamic and survival improvements in CTD-PAH especially in SLE and MCTD [31], but these results have not yet been verified in large prospective RCTs. The 6 Minute Walk Test (6MWT) may be a less useful outcome measure in this population due to peripheral joint pain and other mobility issues. Due to the benefit seen with early treatment [17], screening with annual echo is recommended in those with CTD at risk of developing PAH.
Congenital Heart Disease PAH
Patients with congenital heart disease PAH (CHD-PAH) have an improved prognosis compared with IPAH, with reported 5-year survival of 91 vs 63 %. Four main subgroups exist: Eisenmenger syndrome, PAH associated with systemic to pulmonary shunts, PAH associated with small defects and PAH after cardiac defect correction [32]. The latter group (PAH following cardiac defect correction) have the worst prognosis of the four. Treatment strategies are fundamentally the same as IPAH, with no contraindications to any class of disease-targeted therapy. Survival in Eisenmengers without treatment is superior to survival following lung transplant; therefore, transplantation should be reserved for patients with WHO functional class IV symptoms and estimated 5-year survival of less than 50 % [33].
Porto-Pulmonary Hypertension
Prognosis in porto-pulmonary hypertension (PoPH) is worse, and patients are more likely to be commenced on disease-targeted therapy later than in IPAH [2]. Many RCTs have excluded PoPH from entry; the trials that have included this population have deemed that all classes of PH therapy are efficacious, albeit caution should be exerted with prostacyclins given the bleeding risk and liver function should be monitored closely with ERAs [34]. Calcium channel blockers are relatively contraindicated in PoPH as they cause mesenteric vasodilation, resulting in increased portal pressures [35]. Despite their widespread use in portal hypertension, there is evidence that beta-blockers still negatively impact haemodynamics and exercise capacity in PoPH [34]. Patients who are responsive to vasodilator therapy with reduction in mPAP may benefit from liver transplantation, and the limited evidence that exists suggests that this population has a favourable survival but frequently have ongoing post-transplant pulmonary hypertension requiring treatment [36].
Pulmonary Veno-Occlusive Disease
Prognosis remains exceptionally poor in pulmonary veno-occlusive disease (PVOD) and significantly worse than IPAH, with a lack of effective medical therapy. One-year survival has been reported to be as high as 72 %, even in patients with comparable haemodynamics and walk distance to patients with IPAH, suggesting that standard PAH outcome measures are not accurate in prognosticating PVOD patients. The mainstay of treatment is supportive, with oxygen, diuretics and anticoagulation. Conflicting evidence exists regarding the tolerability and efficacy of disease-targeted therapies, with some small series showing clinical and haemodynamic improvement [37]. All classes, including calcium channel blockers, have been reported to cause acute and severe pulmonary oedema due to arterial vasodilation in the context of venous obstruction. In patients with sarcoid associated PVOD or CTD (excluding scleroderma), there is some evidence suggesting a benefit from immunosuppression [38]. The only curative option for PVOD is lung transplantation, and this should be considered early due to the rapid clinical course and limited treatment options.
Assessing Prognosis and Response to Treatment
Factors that reflect RV function have been shown to correlate with survival, both at baseline and follow-up. More recently, noninvasive surrogate markers of RV function have been explored in assessing prognosis and response to treatment and will be discussed below along with established markers of prognosis.
Aetiology
As mentioned previously, patients with PoPH and CTD-PAH have a significantly poorer outcome compared with IPAH, whereas CHD-PAH patients have an improved prognosis.
Functional Variables and Quality of Life
Functional Class
New York Heart Association (NYHA)/World Health Organisation (WHO) functional class (FC) is considered one of the best predictors of survival [2], despite its interobserver variability [39]. Recently, Nickel et al. demonstrated the utility of FC both at baseline and in response to treatment [40] in a single-centre cohort of 109 patients with IPAH. Patients who were FC III or IV at baseline, who did not improve with treatment, had a significantly worse survival than those who improved to FC I or II, or those who were FC I or II at diagnosis. Furthermore, deterioration in FC during follow-up in this cohort (3–12 months) was associated an increased risk of death. These findings were confirmed in a larger cohort of patients from the REVEAL registry [41].
Quality of Life
Patient-reported symptoms are an important consideration in determining the efficacy of therapy. The Cambridge Pulmonary Hypertension Outcome Review (CAMPHOR) score was predictive of clinical worsening events in a recent retrospective analysis [42]. The emPHasis-10 health-related quality of life questionnaire has been recently developed specifically for PAH. In preliminary analysis, it has been shown to correlate moderately with WHO FC and 6-min walk distance (6mwd), both known independent indicators of prognosis [43].
Exercise Capacity
6-min Walk Distance
Traditionally, the 6-min walk distance has been used as the primary end point in clinical trials assessing the efficacy of new drug therapies [44]. It is an inexpensive, simple, noninvasive test that has been shown to have prognostic value when measured at baseline and also has been found to correlate with baseline haemodyamics reflecting RV function [45]. Its value in assessing response to treatment, however, is limited, particularly in the era of combination therapy, where small changes distance in have been noted in the majority of trials (e.g. SERAPHIN 22 m, STEP 26 m, AMBITION 22 m), and the minimally important difference is approximately 33 m [46]. It is recognised that the 6mwt is less sensitive in fitter subjects. Lee et al. assessed the value of percent-predicted 6mwd derived from currently available reference equations and found that both absolute 6mwd and percent-predicted 6mwd at baseline and during follow-up were predictive of survival, but the percent-predicted value did not provide any greater prognostic information [47].
There may be utility in the 6mwt in follow-up of IPAH patients if additional data such as heart rate recovery (HRR) is analysed. HRR < 16 beats per minute, 1 min after cessation of walking, has been shown to be negatively associated with survival and time to clinical worsening (hazard ratio, 5.2; 95 % CI, 1.8–14.8; p 0.002) [48].
Cardiopulmonary Exercise Testing
Patients with PAH have limited exercise capacity primarily due to an impaired ability to raise stroke volume. A number of other factors contribute such as autonomic dysfunction, peripheral and respiratory muscle weakness. Cardiopulmonary exercise testing (CPET) allows simultaneous assessment of the cardiovascular, metabolic and ventilatory responses to exercise. Low peak oxygen uptake (VO2) measured during incremental CPET has long been established as an independent predictor of survival [49]. Recently, Blumberg et al. demonstrated that VO2 correlated significantly with cardiac index (CI) during exercise (measured by RHC), and both peak VO2 and CI were predictive of survival in PAH patients at baseline [50]. Wensel et al. demonstrated that % peak VO2 provides even greater prognostic information compared with absolute VO2 and is an independent predictor of survival during long-term follow-up [51]. Furthermore, the independent predictors of survival in this study: % predicted peak VO2, change in PVR and change in HR in response to exercise, provided superior prognostic value in combination, than any of the values in isolation, adding to the body of evidence supporting composite trial end points and a multi-faceted approach to assessment of prognosis and response to treatment.
Serial CPET measurements have prognostic utility in determining treatment response and predicting survival. Development of right to left shunt during exercise and a ventilatory equivalent for CO2 (Ve/VCO2) > 40 L min at anaerobic threshold (AT) are significantly associated with worse outcomes [52]. In a single-centre observational study, change in maximal aerobic capacity and chronotropic response at follow-up were significant predictors of subsequent survival in patients undergoing treatment for PAH [53].
Haemodynamic Measurements
Invasive
RHC
All patients must undergo right heart catheterisation to establish the diagnosis of PAH. It can accurately measure pressures and stroke volume but does not provide information on RV volume. The majority of clinical trials have reassessed haemodynamics at 12–16 weeks [54]. Baseline RAP (>15 mmHg), mPAP and mixed venous oxygen saturation are long established markers of survival [3]. Baseline CI > 2.5 L min−1 m−2 is associated with improved survival [40]. A registry analysis of patients undergoing follow-up RHC after 4–29 weeks of treatment, demonstrated short-term improvements in cardiac output of >0.22 L min−1 and a decrease in PVR of >176 dyn s cm−5 were associated with long-term transplant free survival.
Noninvasive
Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance imaging (CMR) provides excellent assessment of RV function, a key determinant of outcome and response to treatment in PAH and is thus considered the gold standard for RV functional assessment. A number of prognostic variables have been studied and full discussion is out with the scope of this article.
The importance of utilising markers directly reflecting RV function was highlighted by Vonk-Noodegraaf et al. Right ventricular ejection fraction (RVEF) measured at baseline was a better predictor of mortality than PVR measured by RHC [55•]. Furthermore, they demonstrated that RV function could still deteriorate despite an improvement in PVR whilst on disease-targeted therapy, and this was associated with a poorer outcome [56•]. Courand et al. showed that patients with baseline RVEF >25 % had improved survival and those with a stable or increased RVEF at 3–6 months had significantly lower cardiovascular mortality [19••].
Van Wolferen et al. demonstrated that a right ventricular end-diastolic volume index (RVEDVI) of <84 mL/m2 at diagnosis had a significantly better survival compared to those with an RVEDVI ≥ 84 mL/m2 [57]. A low SV measured by CMR at baseline was also associated with poor prognosis and displayed greater predictive value than cardiac index (CI). A 10-mL change in stroke volume during follow-up has been suggested as clinically relevant [56•], and a decrease in stroke volume during therapy is an indicator of treatment failure..
In the multi-centre EURO-MR study, Peacock et al. verified these prognostic CMR markers and highlighted the importance of assessing both left-sided and right-sided variables, showing that LV end-diastolic volume (LVEDV) was more closely related to stroke volume than RV end-diastolic volume [58].
Echocardiography
Traditionally, Echocardiography (echo) is used in the initial diagnostic workup of PAH, with a lesser role in follow-up and treatment decisions due to a significant interobserver variability and technical challenges in image acquisition.
Established parameters that relate to poor survival are tricuspid annular plane systolic excursion (TAPSE) (≤15 mm), left ventricular eccentricity index (≥1.7) [59], presence of a pericardial effusion and enlargement of left and right atrial area. Strain and 3-dimensional (3D) RV echo are newer echo modalities that show more promise in assessing response to therapy and predicting prognosis. RV free wall strain has been significantly associated with NYHA FC and long-term survival [60]. Furthermore, 3D echo has been shown to have lower interobserver variability and significantly better agreement with established CMR parameters than traditional 2D echo [61]. There is not yet enough evidence or large-scale experience with 3D or strain echo to incorporate them into current algorithms; however, given the promising results so far, further investigation is warranted.
Inert Gas Rebreathing
Inert gas rebreathing (IGR) has shown promise as a noninvasive, reproducible measure of stroke volume at rest and during exercise. Lee et al. demonstrated that SV measured by IGR during submaximal exercise, detected response to treatment changes in pre-capillary hypertension. Furthermore, in fitter patients, where there are recognised limitations of the 6mwt, SV measured by IGR changed significantly in response to treatment, whereas 6mwd did not [62]. The same group also demonstrated close agreement between SV measured by IGR, CMR and thermodilution at RHC. Further work is required to verify these findings in a larger population.
Serum Biomarkers
Brain natriuretic peptide (BNP) and N-terminal fragment proBNP (NT-proBNP) indirectly reflect RV function and elevated levels correlate with poorer prognosis. Similarly, rising NT-proBNP levels during follow-up are associated with treatment failure and worse prognosis [63]. NT-proBNP rises in chronic renal impairment (CRF); however, it has recently been demonstrated in a population of PAH patients with CRF that NT-pronBNP maintains its value in predicting survival and correlates with haemodynamics [64].
Uric acid [65] and troponin are also associated with disease severity, although do not correlate with RV function.
Newer biomarkers are emerging that may hold future promise as prognostic markers, particularly inflammatory cytokines [66]; however, these are not yet incorporated into current clinical practice.
Composite Risk Scores
Registry data has provided a significant amount of evidence regarding prognosis in PAH. Composite risk scores have been developed from a number of these registries with the largest and most contemparous being the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) registry.
The REVEAL risk calculator [67] estimates the 1-year survival from the point of assessment based on the score obtained from a variety of prognostic markers (Fig. 2). It can be used at any time during the disease course, besides baseline assessment, and has recently been prospectively verified as being predictive of survival and response to treatment [68••].

The reveal registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Benza RL, Gomberg-Maitland M, Miller DP, et al. Chest. 2012;141(2):354–362. doi:10.1378/chest.11-0676. Reproduced with permission. Licence number 3584750153150
Nickel et al. evaluated baseline and follow-up prognostic markers in a cohort of IPAH patients [40] and found four variables were independently associated with survival at baseline and follow-up: NYHA FC I & II, NT-proBNP < 1800 ng/l, SvO2 ≥ 65 % and CI > 2.5 L/min/m
Treatment Algorithm
PAH-targeted therapy is considered in symptomatic patients who are not vasoreactive or who are vasoreactive but display a suboptimal response to treatment with calcium channel blockers (Fig. 3). FC II and III patients should commence one of the orally active therapies whilst parenteral prostanoid is recommended as initial monotherapy for FC IV PAH patients. Despite this recommendation, the REVEAL registry found that a significant proportion of FC IV patients were on oral therapy. Numerous reasons may exist for this, such as the invasive nature of therapy delivery, cost and convenience; however late initiation of parenteral prostanoid is associated with worse outcome, and patients who meet criteria should be commenced on this therapy early [69].

ERS/ECS 2013 Updated treatment algorithm for pulmonary arterial hypertension. Reproduced with permission from original article. The REVEAL Registry Risk Score Calculator in Patients Newly Diagnosed With Pulmonary Arterial Hypertension. RL Benza et al. Chest. 2012 Feb;141(2):354-62. License Number: 3584750153150
Current guidelines base initial treatment recommendations predominantly on functional class at time of diagnosis and recommend initial monotherapy for all, with sequential combination therapy in cases of inadequate clinical response for patients in FC III or IV.
The targeting more than one, a cumulative effect may be produced. There is strong evidence to support sequential combination therapy, with data from meta-analyses showing improved functional class, exercise capacity and haemodynamics, but no mortality benefit [70]. In patients with IPAH and SSc-PAH, the combination of either an ERA or PDE5i with prostanoid appears to be more effective than ERA and PDE5i in combination [71], particularly in those deteriorating on monotherapy.
Comparatively less is known regarding the role of “upfront combination therapy” [72]. The results of the RCT AMBITION, assessing upfront Tadalafil and Ambristenan, are awaited, but initial data appear promising. Attempts have been made to identify those likely to benefit from this strategy retrospectively; Bajwa et al. suggests patients displaying more severe haemodynamics at diagnosis with higher PAH risk score, poorer FC and low oxygen saturation on exercise may be an appropriate cohort; however, this needs prospectively verified and objectified on a larger scale [73]. Upfront combination therapy is likely to have a role in the future for severe PAH and possibly for young fitter patients in whom a more aggressive treatment stance may be taken. This approach is likely to affect clinician preference in initial drug choice within a class, as certain combinations seem to be more effective [71]. Further research should focus on PAH subgroups to identify the most appropriate timings and combinations for specific patient populations.
Small, single-centre case reports also exist of triple upfront combination therapy use in PAH, with improved survival, haemodynamics and functional class [74]; however, caution should be exerted in drawing conclusions due to lack of randomised, robust evidence.
Goal-Directed Therapy
Goal-directed therapy is a relatively novel advance in the management of PAH, being first described by Hoeper et al. in 2005 [75] with the aim of improving long-term survival. This strategy has proved effective in reducing the need for IV prostacyclin and referral for lung transplant. The main difference between goal-directed therapy and the previous standard treatment approach is that a patient may be stable or show a slight improvement on disease-targeted therapy, but not fulfil treatment goals, and thus warrant treatment escalation under current recommendations. No single parameter can fulfil the role of a reliable prognostic indicator; therefore, multiple variables known to reflect prognosis are assessed on a 3–6 monthly basis to determine treatment response. The ERS /ECS revised their recommended goals in 2013; the following treatment goals are advised to assess efficacy of therapy and trigger treatment escalation (Table 1) [76••].
Conclusions
Significant advances have been made in the medical management and understanding of pulmonary arterial hypertension in the past decade. Outlook is significantly better and composite prognostic markers now focus on longer-term rather than short-term outcomes. A wider variety of drug treatments are available, and the initial choice of agent is based on the disease severity at diagnosis, patient preference including route of administration, side effect profile and future therapy escalation plans, taking into consideration the efficacy of particular drug combinations. It is likely that upfront combination therapy will be incorporated into future guidelines, but further data are needed to verify and strengthen this recommendation.
Further research should focus on establishing more convenient, tolerable routes for prostacyclin therapy, determining the optimal timing and role for combination therapy and investigating novel pharmacological targets and compounds.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Simonneau G et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34–41.
Benza RL et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010;122(2):164–72.
D'Alonzo GE et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115(5):343–9.
Humbert M et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122(2):156–63.
Wolfson AM, Steiger N, Gomberg-Maitland M. New pharmacotherapies for pulmonary hypertension: where do they fit in? Curr Hypertens Rep. 2014;16(12):496.
Galie N et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60–72.
Buckley MS, Feldman JP. Inhaled epoprostenol for the treatment of pulmonary arterial hypertension in critically ill adults. Pharmacotherapy. 2010;30(7):728–40.
Benza RL et al. One-year experience with intravenous treprostinil for pulmonary arterial hypertension. J Heart Lung Transplant. 2013;32(9):889–96.
Lopez-Medrano F et al. High incidence of bloodstream infection due to gram-negative bacilli in patients with pulmonary hypertension receiving intravenous treprostinil. Arch Bronconeumol. 2012;48(12):443–7.
Steringer-Mascherbauer R et al. Intravenous treprostinil delivered by the implantable pump Lenus Pro®: a innovative “surgical” approach to management of PAH. J Heart Lung Transplant. 2013;32(4):S64.
Buckley MS et al. Clinical utility of treprostinil in the treatment of pulmonary arterial hypertension: an evidence-based review. Core Evid. 2014;9:71–80.
Olschewski H et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322–9.
Galie N et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39(9):1496–502.
Simonneau G et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J. 2012;40(4):874–80.
Galie N et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010–9.
Oudiz RJ et al. Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(21):1971–81.
Galie N et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008;371(9630):2093–100.
McGoon MD et al. Ambrisentan therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function test abnormalities. Chest. 2009;135(1):122–9.
Pulido T et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–18. Macitentan demonstrated safe, efficacious results especially in relation to liver function tests. This trial also demonstrated improved survival and morbidity with Macitentan as combination therapy with PDE5 inhibitors, where other trials assessing PDE5 and ERAs were less promising.
Galie N et al. Liver toxicity of sitaxentan in pulmonary arterial hypertension. Eur Heart J. 2011;32(4):386–7.
Perez-Penate GM et al. Long-term inhaled nitric oxide plus phosphodiesterase 5 inhibitors for severe pulmonary hypertension. J Heart Lung Transplant. 2008;27(12):1326–32.
Barst RJ et al. Clinical perspectives with long-term pulsed inhaled nitric oxide for the treatment of pulmonary arterial hypertension. Pulm Circ. 2012;2(2):139–47.
Wang RC et al. Efficacy and safety of sildenafil treatment in pulmonary arterial hypertension: a systematic review. Respir Med. 2014;108(3):531–7.
Oudiz RJ et al. Tadalafil for the treatment of pulmonary arterial hypertension: a double-blind 52-week uncontrolled extension study. J Am Coll Cardiol. 2012;60(8):768–74.
Lichtblau M et al. Safety and long-term efficacy of transition from sildenafil to tadalafil due to side effects in patients with pulmonary arterial hypertension. Lung. 2014;192(6):947–54.
Jing ZC et al. Vardenafil in pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled study. Am J Respir Crit Care Med. 2011;183(12):1723–9. Macitentan demonstrated safe, efficacious results especially in relation to liver function tests. This trial also demonstrated improved survival and morbidity with macitentan as combination therapy with PDE5 inhibitors, where other trials assessing PDE5 and ERAs were less promising.
Wirostko BM et al. Ocular safety of sildenafil citrate when administered chronically for pulmonary arterial hypertension: results from phase III, randomised, double masked, placebo controlled trial and open label extension. BMJ. 2012;344:e554.
Rubin LJ, et al. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2). Eur Respir J. 2015.
Hoeper MM et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127(10):1128–38.
Le Pavec J et al. Systemic sclerosis-related pulmonary hypertension associated with interstitial lung disease: impact of pulmonary arterial hypertension therapies. Arthritis Rheum. 2011;63(8):2456–64.
Miyamichi-Yamamoto S et al. Intensive immunosuppressive therapy improves pulmonary hemodynamics and long-term prognosis in patients with pulmonary arterial hypertension associated with connective tissue disease. Circ J. 2011;75(11):2668–74.
Manes A et al. Current era survival of patients with pulmonary arterial hypertension associated with congenital heart disease: a comparison between clinical subgroups. Eur Heart J. 2014;35(11):716–24.
Krishnan U, Rosenzweig EB. Pulmonary arterial hypertension associated with congenital heart disease. Clin Chest Med. 2013;34(4):707–17.
Porres-Aguilar M, Mukherjee D. Portopulmonary hypertension: an update. Respirology. 2015;20(2):235–42.
Ota K et al. Effects of nifedipine on hepatic venous pressure gradient and portal vein blood flow in patients with cirrhosis. J Gastroenterol Hepatol. 1995;10(2):198–204.
Khaderi S et al. Long-term follow-up of portopulmonary hypertension patients after liver transplantation. Liver Transpl. 2014;20(6):724–7.
Ogawa A et al. Safety and efficacy of epoprostenol therapy in pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. Circ J. 2012;76(7):1729–36.
Huertas A et al. Pulmonary veno-occlusive disease: advances in clinical management and treatments. Expert Rev Respir Med. 2011;5(2):217–29. quiz 230–1.
Taichman DB, Mcgoon MD, Harhay MO, Archer-Chicko C, Murugappan M, Chakinali MM, et al. Wide variation in clinician assessment of who fc 2009.pdf. Mayo Clin Proc. 2009;84(7):586–92.
Nickel N et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2012;39(3):589–96.
Barst RJ et al. Functional class improvement and 3-year survival outcomes in patients with pulmonary arterial hypertension in the REVEAL Registry. Chest. 2013;144(1):160–8.
McCabe C et al. Patient-reported outcomes assessed by the CAMPHOR questionnaire predict clinical deterioration in idiopathic pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Chest. 2013;144(2):522–30.
Yorke J et al. emPHasis-10: development of a health-related quality of life measure in pulmonary hypertension. Eur Respir J. 2014;43(4):1106–13.
Gomberg-Maitland M et al. New trial designs and potential therapies for pulmonary artery hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D82–91.
Miyamoto S et al. Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension. Comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med. 2000;161(2 Pt 1):487–92.
Mathai SC et al. The minimal important difference in the 6-minute walk test for patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186(5):428–33.
Lee WT, Peacock AJ, Johnson MK. The role of per cent predicted 6-min walk distance in pulmonary arterial hypertension. Eur Respir J. 2010;36(6):1294–301.
Minai OA et al. Heart rate recovery predicts clinical worsening in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185(4):400–8.
Wensel R. Assessment of survival in patients with primary pulmonary hypertension: importance of cardiopulmonary exercise testing. Circulation. 2002;106(3):319–24.
Blumberg FC et al. Impact of right ventricular reserve on exercise capacity and survival in patients with pulmonary hypertension. Eur J Heart Fail. 2013;15(7):771–5.
Wensel R et al. Incremental prognostic value of cardiopulmonary exercise testing and resting haemodynamics in pulmonary arterial hypertension. Int J Cardiol. 2013;167(4):1193–8.
Oudiz RJ et al. Usefulness of right-to-left shunting and poor exercise gas exchange for predicting prognosis in patients with pulmonary arterial hypertension. Am J Cardiol. 2010;105(8):1186–91.
Groepenhoff H et al. Prognostic relevance of changes in exercise test variables in pulmonary arterial hypertension. PLoS One. 2013;8(9):e72013.
Tiede H et al. Short-term improvement in pulmonary hemodynamics is strongly predictive of long-term survival in patients with pulmonary arterial hypertension. Pulm Circ. 2013;3(3):523–32.
van de Veerdonk MC et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol. 2011;58(24):2511–9. This highlights the prognostic value of haemodynamic factors in prognostication and assessing treatment response. PVR could fall on treatment but patients could still deteriorate or die. RV ejection fraction correlated with survival.
van Wolferen SA et al. Clinically significant change in stroke volume in pulmonary hypertension. Chest. 2011;139(5):1003–9. This identifies a minimally important difference for RV ejection fraction: a key prognosticator. This will greatly aid treatment decisions and end points in future clinical trials.
van Wolferen SA et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J. 2007;28(10):1250–7.
Peacock AJ et al. Changes in right ventricular function measured by cardiac magnetic resonance imaging in patients receiving pulmonary arterial hypertension-targeted therapy: the EURO-MR study. Circ Cardiovasc Imaging. 2014;7(1):107–14.
Ghio S et al. Prognostic relevance of the echocardiographic assessment of right ventricular function in patients with idiopathic pulmonary arterial hypertension. Int J Cardiol. 2010;140(3):272–8.
Blanchard DG, DeMaria AN. Right ventricular 3-dimensional strain in pulmonary hypertension: the quest to see the future. J Am Coll Cardiol. 2014;64(1):52–3.
Jenkins C et al. Reproducibility of right ventricular volumes and ejection fraction using real-time three-dimensional echocardiography: comparison with cardiac MRI. Chest. 2007;131(6):1844–51.
Lee WT et al. Use of non-invasive haemodynamic measurements to detect treatment response in precapillary pulmonary hypertension. Thorax. 2011;66(9):810–4.
Souza R et al. NT-proBNP as a tool to stratify disease severity in pulmonary arterial hypertension. Respir Med. 2007;101(1):69–75.
Harbaum L et al. N-terminal pro-brain natriuretic peptide is a useful prognostic marker in patients with pre-capillary pulmonary hypertension and renal insufficiency. PLoS One. 2014;9(4):e94263.
Nagaya N et al. Serum uric acid levels correlate with the severity and the mortality of primary pulmonary hypertension. Am J Respir Crit Care Med. 1999;160(2):487–92.
Cracowski JL et al. Proinflammatory cytokine levels are linked to death in pulmonary arterial hypertension. Eur Respir J. 2014;43(3):915–7.
Benza RL et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012;141(2):354–62.
Benza RL, et al. Prognostic implications of serial risk score assessments in patients with pulmonary arterial hypertension: a registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL) analysis. J Heart Lung Transplant. 2014. Confirmed the need for serial, composite risk stratification in assessment of prognosis and treatment response as opposed to utilising single variables such as 6mwd.
Badagliacca R et al. Prognostic factors in severe pulmonary hypertension patients who need parenteral prostanoid therapy: the impact of late referral. J Heart Lung Transplant. 2012;31(4):364–72.
Fox BD, Shimony A, Langleben D. Meta-analysis of monotherapy versus combination therapy for pulmonary arterial hypertension. Am J Cardiol. 2011;108(8):1177–82.
Johnson SR et al. Dual therapy in IPAH and SSc-PAH. A qualitative systematic review. Respir Med. 2012;106(5):730–9.
Ghofrani HA, Humbert M. The role of combination therapy in managing pulmonary arterial hypertension. Eur Respir Rev. 2014;23(134):469–75.
Bajwa AA, et al. Predicting the need for upfront combination therapy in pulmonary arterial hypertension. J Cardiovasc Pharmacol Ther. 2015.
Sitbon O et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J. 2014;43(6):1691–7.
Hoeper MM et al. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur Respir J. 2005;26(5):858–63.
McLaughlin VV et al. Treatment goals of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D73–81. The most recent guidelines incorporate goal-directed therapy and much more ambitious composite treatment goals. This signifies a change in treatment strategy—patients stable, but not meeting these new goals would previously have been maintained on monotherapy, but now more aggressive treatment with combination therapy is advocated.
Compliance with Ethics Guidelines
Conflict of Interest
Andrew J. Peacock declares honoraria, assistance with travel to meetings and unrestricted research grants from Actelion, Bayer, Gilead, GSK and United Therapeutics. Alison M. MacKenzie declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Pulmonary Hypertension
Rights and permissions
About this article

Cite this article
MacKenzie, A.M., Peacock, A.J. Medical Therapies for the Treatment of Pulmonary Arterial Hypertension: How Do We Choose?. Curr Hypertens Rep 17, 56 (2015). https://doi.org/10.1007/s11906-015-0560-2
Published:
DOI: https://doi.org/10.1007/s11906-015-0560-2