
Abstract
Purpose of the Review
Tricuspid regurgitation is associated with increased mortality in proportion to right ventricular adaptation to increased volume loading and pulmonary artery pressure. We here review recent progress in the understanding of right ventricular adaptation to pre- and after-loading conditions for improved recommendations of tricuspid valve repair.
Recent Findings
Trans-catheter tricuspid valve repair has made the correction of tricuspid regurgitation more easily available, triggering a need of tighter indications. Several studies have shown the feasibility and relevance to the indications of tricuspid valve repair of imaging of right ventricular ejection fraction measured by magnetic resonance imaging or 3D-echocardiography, and the 2D-echocardiography of the tricuspid annular plane systolic excursion to systolic pulmonary artery pressure ratio combined with invasively determined mean pulmonary artery pressure and pulmonary vascular resistance.
Summary
Improved definitions of right ventricular failure and pulmonary hypertension may be considered in future recommendations on the treatment of tricuspid regurgitation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
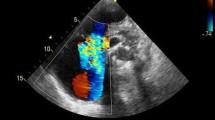
Tricuspid regurgitation is an independent predictor of mortality in proportion to its severity [1]. The negative impact of tricuspid regurgitation on survival is worsened by coexistent right ventricular (RV) function alterations and increased pulmonary artery pressure (PAP) [2]. The reasons why tricuspid regurgitation alters RV function adaptation to loading and decreases survival are not entirely understood. However, the culprit is chronically increased volume loading of the RV by the adding up of regurgitated flow to systemic venous return [3••, 4••]. Accordingly, tricuspid regurgitation can be basically considered as a model of chronic RV volume overload. This is illustrated in Fig. 1, which shows a 2-D echocardiographic 4-chamber view of dilated right heart chambers and massive tricuspid regurgitation.

Two-dimensional echocardiography with an apical 4-chamber view of a dilated right heart and color Doppler demonstration of massive tricuspid regurgitation
We here review recent progress in the pathophysiology of RV volume overload, with or without increased afterload and its clinical relevance in the particular context of tricuspid regurgitation.
Right Ventricular Function Adaptation to Volume Versus Pressure Loading
The right ventricle (RV) in mammals and in birds is a thin-walled crescent shape structure coupled to systemic venous return on one side and to the pulmonary circulation on the other side. Low pressures in the pulmonary circulation preserves the integrity a thin and fragile blood-gas barrier designed for high levels of gas exchange in endothermic animals [5]. In fact, pulmonary vascular resistance (PVR) is normally so low that RV does not have much to add to mean systemic filling pressure to drive venous return through the pulmonary circulation to the left heart. In 1943, Starr and his colleagues showed that ablation of the RV in dogs is compatible with life with little change in systemic venous pressures, supporting a notion of a “dispensable” RV [6]. This was repeatedly confirmed, offering background to the introduction of cavo-pulmonary anastomosis as a palliative intervention for certain cardiac malformations, in 1971 by Fontan and Baudet [7]. Tricuspid regurgitation would not matter to a dispensable RV. This may be the main reason for the excellent long-term clinical tolerance to an isolated tricuspid regurgitation in sedentary healthy subjects.
Patients with the so-called Fontan circulation enjoy a normal life without RV for several decades, but may rapidly deteriorate in case of increased PAP on, for example altitude exposure or on increased left ventricular (LV) filling pressures [8]. High level aerobic exercise may be another cause of increased PAP but mainly related to an increased cardiac output [9]. The contribution of RV pump function to maximum cardiac output is indirectly shown by a 30 to 50% decrease in predicted maximum oxygen uptake in healthy young Fontan patients [10]. From these observations, it can be inferred that a normal RV is required for an adapted cardiac output response to increased metabolic demand of exercise. As exercise is associated with an increased systemic venous return, additional volume loading due to tricuspid regurgitation could be functionally deleterious. This may be a reason of decreased exercise capacity in healthy subjects with an isolated tricuspid regurgitation, even though there is no reported data to show it.
The structure of the RV is not designed to cope with brisk increases in PAP. In anesthetized animals, pulmonary ensnarement associated with a maximum systolic RV pressure exceeding 60 mmHg, which corresponds to mean PAP (mPAP) of 30–35 mmHg, is associated with extreme RV dilatation, a circulatory collapse and cardiac arrest [11]. However, if given time, the RV adapts to increased loading by an increased contractility. This “homeometric adaptation” is turned on within 3 to 5 min, and is not therefore dependent on hypertrophic remodeling. In Starling’s heart–lung preparation, an acute increase of either preload or afterload is immediately associated with an increase in end-diastolic and end-systolic volumes (EDV and ESV), a decrease ejection fraction (EF) and a preserved stroke volume (SV) [12]. But after several minutes, this “heterometric adaptation” conforming to so-called Starling’s law of the heart is replaced by a homeometric adaptation also called Anrep’s law of the heart as EDV returns to normal, ESV is decreased, EF increases and SV is back to baseline [13]. Starling was worried that these observations were due to a deterioration of the hear-lung preparation over time and argued with his then student Anrep about it. However, it is now better understood that Starling's law of the heart applies to beat-by-beat changes in loading, or when the homeometric adaptation eventually fails. A normal RV adapts to prolonged volume loading with flow output matching venous return without increased dimensions (Fig. 2). This may be another basic reason for good tolerance to increased venous return situations including exercise, in healthy subjects with isolated tricuspid regurgitation.

Time-course of RV volume changes after a brisk increase in venous return in Starling’s heart–lung preparation observed during 10 min. The initial heterometric adaptation described by Starling et al. is followed by a homeometric adaptation allowing for a return to initial end-diastolic volume (EDV) with decreased end-systolic volume (ESV) and increased stroke volume (EDV-ESV). From ref 13 by Rosenblueth A, Alanis J, Lopez E, Rubio R. The adaptation of ventricular muscle to different circulatory conditions. Arch Int Physiol Biochim. 1959; 67: 358–373
Contractility responses to increased preload or afterload vary in magnitude depending on time course of increased loading, volume status, reactive ventricular hypertrophy, the presence or not of systemic diseases and yet unknown differences in molecular signaling pathways [3••, 4••, 14]. When a homeometric adaptation gets exhausted, the heart is returned to a heterometric adaptation but this may be at the price of increased dimensions and filling pressures, and appearance at some point of signs of systemic congestion [3••, 4••, 14].
In tricuspid regurgitation, the RV is constrained into an initial heterometric adaptation to volume loading, with adjustment of its coupling to the pulmonary circulation [3••, 4••]. The regurgitated volume during systole constantly prevents the return of diastolic volumes to normal. Therefore more enhancement of systolic function may be required to prevent excessive RV dilatation.
Right Ventricular-Arterial Coupling
The adaptation of RV function to loading conditions is essentially systolic, with constant adjustment of contractility to afterload. The gold standard measure of contractility is end-systolic elastance (Ees), or the ratio between and systolic pressure (ESP) and ESV. The gold standard measurement of afterload is arterial elastance (Ea), or the ratio between SV and ESV. There is an optimal ratio of Ees/Ea of approximately 2 allowing for blood flow ejection from the RV into the pulmonary circulation at a minimal energy cost [3••, 14]. The system has reserve, as in pulmonary hypertension the Ees/Ea may decrease by 50% before the RV dilates by heterometric adaptation [15].
It may be noted that Ees and Ea have a common pressure term, so that the ratio can be simplified as SV/ESV [16] which in turn is equal to EF/(1-EF) [17]. Simple modeling predicts that both these volume surrogates of Ees/Ea can decrease as well by some 50%, with cut-off values around 35% for EF and 54% for SV/ESV before RV dilatation occurs [17]. This has been confirmed for RVEF [15].
Measurements of Ees and Ea require conductance catheter technology for high-fidelity instantaneous measurements of pressure and volumes. This is expensive and requires sophisticated expert catheterization laboratory environment. Unsurprisingly therefore, reported measurements are limited to only small patient cohorts, mainly with severe pulmonary hypertension, with no study specifically addressing volume overload, and even less so tricuspid regurgitation.
It has been previously assumed that the Ees/Ea ratio would be initially preserved in volume overload [3••]. Typical RV pressure–volume relationships for the determination of Ees and Ea in a healthy subject, a patient with severe pulmonary hypertension and a patient with volume overload are shown in Fig. 3. It does not predispose a homeometric reversal of RV enlargement illustrated in Fig. 2. Yet, it may apply to patients with tricuspid regurgitation-related permanent increase in RV volumes, even if then one would conceive a higher than normal Ees to cope with inevitable increase blood volume to eject.

Graphical representation of RV pressure–volume relationships in a normal RV, a volume overloaded RV and, for comparison, a pressure overloaded RV. Note that in both RV volume and pressure overload there is a rightward shift of the loops reflecting RV dilatation. However, contractility (end-systolic elastance) does not differ between the normal and the volume overloaded RV, whereas it is significantly increased in the pressure overloaded RV to match pulmonary arterial load (arterial elastance) and maintain ventriculoarterial coupling. There is mild increase in the end-diastolic pressure–volume relationship in volume overload, but less prominent than in pressure overload. Ea: arterial elastance; Eed: end-diastolic elastance; Ees: end-systolic elastance; EDPVR: end-diastolic pressure–volume relationship; EDV: end-diastolic volume; ESV: end-systolic volume; Pes: end-systolic RV pressure; Pmax: maximal RV pressure. Modified from ref 3 Sanz J, Sanchez-Quintana D, Bossone E, Bogaard HJ, Naeije R. Anatomy, function, and dysfunction of the right ventricle: JACC State-of-the-Art Review. J Am Coll Cardiol 2019; 73: 1463–1482, with permission
Anatomic and Functional Adaptation to RV Volume Overload
From a macroscopic perspective, the anatomic hallmarks of isolated or predominant chronic RV volume overload typically include RV enlargement, normal RV wall thickness (but with an increase in free wall mass secondary to dilatation), and predominantly diastolic septal flattening with concomitant reduction in left ventricular (LV) diastolic volume. These features are illustrated in Fig. 4.

Visualization of diastolic flattening of the interventricular septum in a patient with volume overloaded right ventricle. A M-mode echocardiography in parasternal long axis view detects motion patterns of the interventricular septum (IVS) and the inferolateral left wall (ILW). ECG is depicted in green accordingly. Of note, while at the onset of diastole the ILW begins to move away from the left ventricular cavity (blue arrow, line 1), whereas the interventricular septum continues to move towards the left ventricular cavity (between lines 1 and 2) before the motion away from the ventricular cavity begins. B Parasternal short-axis view in end-diastole demonstrates a flattened interventricular septum as a sign of right ventricular volume overload in right cardiac failure. Arrowhead indicates pericardial effusion. C: Reduced distance between interventricular septum and infero-lateral wall in early diastole coincides with prolonged interventricular septal motion towards the ventricular cavity (compare to A: phase between lines 1 and 2)
In addition to 2D echocardiography, imaging modalities of RV volume overload also include 3D echocardiography, cardiac magnetic resonance (CMR), computed tomography (CT) and, to a limited extent, nuclear techniques such as single photon emission computed tomography (SPECT) [18, 19].
Echocardiography is by far the most widely available and can provide important information regarding diastolic function and hemodynamics. CMR is considered the gold standard for quantification of biventricular size and systolic function. CT is a good (although irradiating) alternative to both in the event they are non-diagnostic or they cannot be performed.
Volume overload of the RV is often believed to course with preserved systolic function [3••]. This notion has been supported by in vitro observations of preserved contractility of RV and LV cardiomyocytes from chronically volume overloaded experimental animals [20] and generally good long-term clinical tolerance of isolated mild-to-moderate tricuspid regurgitation such as for example in patients with Ebstein’s anomaly [21••, 22••].
However, in vivo experimental studies show that chronic volume overload is eventually associated with irreversible RV function deterioration. Models of aorta-cava shunt or pulmonary regurgitation are associated with persistent RV dilatation and fibrosis and impaired contractility responses, depending on duration and severity of shunting or regurgitation [23]. Conductance catheter studies in dogs with chronic arterio-venous shunts showed preserved or slightly increased RV Ees, but insufficiently so to prevent increased end-diastolic pressure and volume, suggesting insufficient homeometric adaptation [24]. Magnetic resonance and conductance catheter measurements in pigs after 3 months of experimental pulmonary regurgitation demonstrated reduced baseline RVEF with no significant increase with dobutamine, and a normal Ees that was unresponsive to an inotropic challenge, suggesting an intrinsic alteration in contractility [25]. In dogs with experimental tricuspid regurgitation, RV pressure–volume loops showed a flattening with decreased ejection pressures and greatly increased EDV, so that assessment of Ees adaptation to markedly decreased Ea appeared difficult [26]. The authors therefore defined RV performance by stroke work vs EDV relationships, which turned-out to be markedly depressed. The study also disclosed a marked decrease in a bi-ventricular alteration of β-adrenergic receptor signaling [26]. In pigs with experimental pulmonary regurgitation and altered RV function, the only predictor of functional non-recovery after percutaneous pulmonary valve replacement was the degree of RV dilatation defined an EDV > 120 ml/m2 and an ESV > 45 ml/m2 [27].
With respect to human data, studies of RV volume overload have consistently shown the features indicated above of RV dilatation with abnormal septal motion and decreased LV dimensions (Fig. 4) [28,29,30]. In an early pioneer investigation evaluating RV pressure–volume relationships invasively, the shape of the loops was indistinguishable from that of the normal RV in 5 young patients (median age 14 years) with RV volume overload secondary to an atrial septal defect [31]. However, a study in patients with pulmonary regurgitation due to dysfunctional RV to pulmonary artery conduits who underwent percutaneous pulmonary valve implantation suggested limited contractile reserve as demonstrated by lack of improvement in cardiopulmonary exercise testing after intervention. The authors hypothesized that the volume overloaded RV is already working at a high level at rest with limited ability to increase performance [32].
Impaired contractile reserve related to RV distension has also been demonstrated in patients with pulmonary hypertension and preserved RV-PA coupling [33]. In these patients, volume loading caused a precipitous decrease in the Ees/Ea, which was correlated not only to increased PAP, but also to increased EDV. This is illustrated in Fig. 5, which represents RV pressure–volume loops in a patient with pulmonary hypertension and in a healthy control. Thus experimental animal and human data concur to the notion that RV distension by either initial volume overload or heterometric adaptation to chronically increased afterload is associated with eventual insufficient homeometric adaptation and RV-PA uncoupling.

Families of right ventricular pressure–volume loops at decreasing venous return to define end-systolic and arterial elastances (Ees and Ea) before (in blue) and after fluid loading (MVL) (in orange) in a patient with severe pulmonary hypertension (A) and in a control (B). Fluid loading was associated with a marked decrease in Ees/Ea in the pulmonary hypertensive patient, indicating decreased contractile reserve. The Ees/Ea ratio was unchanged by fluid loading in the control subject. After ref 3 by Kremer N, Rako Z, Douschan P, Gall H, Ghofrani HA Grimminger F et al. Unmasking right ventricular-arterial uncoupling during fluid challenge in pulmonary hypertension. J Heart Lung Transplant 2022; 41: 345–355, with permission
Ventricular Interactions
An important consequence of RV volume overload is its effects on LV geometry and performance because of ventricular interdependence [34]. The RV and LV are intimately interrelated through the inter-ventricular septum, the pericardium, shared coronary blood flow and epicardial circumferential myocyte bundles. Experimental and clinical studies demonstrate that dilatation in one ventricle leads to decrease in size and an upward shift of the pressure–volume curves in the other, thus leading to higher diastolic pressures for the same volume. This is illustrated in Fig. 6 by LV pressure–volume loops in a patient with heart failure and preserved ejection fraction before and after trans-catheter tricuspid valve repair for severe symptomatic tricuspid regurgitation [35].

Pressure–volume loops of the left ventricle (LV) before (in red) and after (in blue) transcatheter tricuspid valve repair (TTVR) in a patient with heart failure and preserved ejection fraction. The procedure improved LV diastolic compliance and filling and increased stroke volume by improved diastolic ventricular interaction. From reference 35by Kresoja KP, Rommel KP, Thiele H, Lurz P. Ventricular interaction in a patient with heart failure with preserved ejection fraction and severe tricuspid regurgitation. Circ Heart Fail 2021; 14: 2021;14:e008768, with permission
This diastolic interdependence is a cause of altered diastolic function and under-filling of the LV because of increased RV dimensions, which may eventually lead to atrophic changes and depressed systolic function of so-called "shrinking LV" (34). The primary mechanism for these changes is LV under-filling due predominantly to septal displacement and changes in LV geometry as illustrated in Fig. 4
Cellular and Molecular Adaptation to RV Volume Overload
The RV response to pressure overload has been extensively characterized in recent reviews [3••, 4••, 14, 36]. One of the main features is cardiomyocyte hypertrophy, which occurs through accumulation of sarcomeric proteins and is usually accompanied by the re-emergence of a fetal gene expression pattern with an increased expression of natriuretic peptides and a “proteomic switch” from α- to β-myosin heavy chain. The advantage of the β-myosin heavy chain is that it exhibits reduced energy requirements, but at the cost of decreased contractility [37]. In addition, there is experimental and some human evidence of a “metabolic switch” from mitochondrial oxidation of fatty acids or glucose as the main mechanism for energy production to increased cytosolic aerobic glycolysis, which has lower oxygen requirements but also reduced energy efficiency [38]. Other changes actually similar to those observed in LV adaptation to systemic hypertension include interstitial fibrosis, inflammatory infiltrates, activation of growth factors, increased pro-inflammatory and pro-apoptotic signaling, disseminated cardiomyocyte apoptosis and areas of focal necrosis [3••, 4••, 14, 36].
The cellular and molecular mechanisms of RV adaptation to volume overload are less well established, but appear to be qualitatively similar though with less hypertrophic and fibrotic changes [23].
A small study in 11 patients with atrial septal defect (ASD) evaluated with PET and SPECT demonstrated an increased uptake of glucose in the inter-ventricular septum compared to the LV free wall, and a similar trend in the RV free wall that however did not reach statistical significance. No differences between the septal and LV free walls were noted for the uptake of thallium as a surrogate of ß-methyl-p-iodophenylpentadecanoic acid, a fatty acid analogue [39]. The increased glucose metabolic rate might represent metabolic switch, although other explanations may be invoked such as false positives on technical problems, increased septal contribution to RV ejection and ischemia [40].
Right ventricular hypertrophy and increased afterload can be associated with increased oxygen requirements and relative ischemia [41]. This has been well documented in RV pressure overload with associated right heart failure. However, smaller degrees of increased wall tension in RV volume overload may be sufficient to cause imbalance in oxygen supply/demand, which in turn might contribute to ischemia-related fibrosis. In the normal RV, and as opposed to the LV, there is substantial coronary blood flow during systole [42], which in theory would make systolic (pressure) overload more susceptible to ischemia than diastolic (volume) overload.
Clinical Course and Management of RV Volume Overload
Because of the above discussed anatomic and physiological features, chronic volume overload of the RV is better tolerated than pressure overload, and patients may be clinically and hemodynamically compensated for decades [21••]. However, chronic volume overload eventually leads to increased morbi-mortality, particularly in the presence of superimposed pressure overload or marked RV enlargement and/or systolic dysfunction [21••]. This evolves to clinical heart failure with shortness of breath, edema, abdominal bloating, fatigue or palpitations in relation to elevated central venous pressure, hepatic congestion, and reduced output or arrhythmia, particularly atrial fibrillation. It must be noted that if volume overload is associated pulmonary hypertension, or leads to, elevated pulmonary pressures (i.e., systemic-to-pulmonary shunts), symptoms such as dyspnea, chest pain, or near syncope may predominate over those of systemic congestion.
The basis of clinical management for RV volume overload is serial imaging to evaluate for progressive RV dilatation and/or systolic function that would trigger surgical or percutaneous repair of the underlying responsible lesion [4••, 21••, 22••]. There is limited role of medical therapy with two main exceptions. In advanced, clinically manifest right heart failure, diuretics (typically loop diuretics and/or aldosterone antagonists) may help alleviate systemic venous congestion [4••, 21••]. The second situation is when pulmonary hypertension coexists or develops during disease course, in which case therapies targeting the pulmonary circulation acting on prostacyclin, nitric oxide, and endothelin signaling pathways may also play a role [43]. In addition, specific etiologies of valvular dysfunction may require dedicated medical therapy, for example antibiotics in infective endocarditis [4••, 21••].
The management of RV volume overload is guided by its initial etiology, which may or not be correctable. Isolated or predominant RV volume overload can be secondary to right-sided regurgitant valve disease and pre-tricuspid systemic-to-pulmonary shunts. Valvular regurgitation can be classified as primary if the abnormality leading to insufficiency resides in the valve itself, or secondary (or functional) when it develops as a result of RV or pulmonary artery dilatation in the setting of other processes such as pulmonary hypertension. In the latter case, the valvular abnormality cannot be seen as the trigger of RV volume overload, but it can contribute to its magnification. Congenital abnormalities with sufficient pre-tricuspid systemic-to-pulmonary shunting can also cause RV volume overload. This therefore includes an ASD or partial anomalous pulmonary venous return. While ventricular septal defects also cause increased volume to the RV, the higher-pressure component of the shunt leads to faster development of pulmonary hypertension, and thus combined pressure and volume overload [43]. Abnormal pulmonary venous return involving only one vein without an associated ASD typically does not cause enough left-to-right shunting to justify significant RV overload or therapeutic intervention [21••].
Tricuspid Regurgitation
Tricuspid regurgitation can be caused by intrinsic valvular abnormalities of various etiologies such as prolapse, radiation, endocarditis, and others. However, functional tricuspid regurgitation secondary to annular dilatation and/or distortion, most commonly in the setting of RV enlargement, represents 80% of cases and is often associated with left-sided valvular or ventricular abnormalities and/or pulmonary hypertension [21••, 22••]. In these cases, tricuspid regurgitation may contribute to additional overload and a spiral of progressive RV dilatation, systolic dysfunction and further regurgitation [44].
It is well known that the presence of severe tricuspid regurgitation is related to poor prognosis in the long term, which has been mostly studied in the context of chronic heart failure or left-sided valvular disease [22••], but also in isolated tricuspid regurgitation [45]. It has been previously thought that the excess mortality associated with severe tricuspid regurgitation is independent from RV dilatation and systolic dysfunction [46]. This notion was supported by both early echocardiographic [1] and more recent echocardiographic and CMR data [47, 48]. However, not all studies concur [2, 49]. How RV status modulates the clinical natural history of tricuspid regurgitation is not fully understood. Nonetheless, its evaluation may be helpful for the timing of interventions.
Imaging studies have attempted at the definition criteria of irreversible RV failure as a contra-indication to valve replacement. In 69 patients undergoing surgical repair of severe residual tricuspid regurgitation after prior left-sided valve surgery, a RV end-systolic area < 20 cm2 as measured by echocardiography predicted reduced risk of death and cardiovascular re-hospitalization [50]. Analogous data were reported in 75 patients evaluated with CMR and in whom a preoperative RVEF ≥ 45%) or an RV end-systolic volume ≤ 75 ml/m2 were linked to improved survival [51]. In 32 patients undergoing surgical repair of residual functional tricuspid regurgitation following previous left-sided valve surgery, a threshold of RV end-diastolic volume < 164 ml/m2 by CMR could identify those demonstrating normal postoperative RVEF [51]. A RVEF lower than 45% measured at 3D echocardiography in another study was associated with an increased mortality of valve replacement in 75 patients with trans-catheter tricuspid regurgitation [52]. Altogether, these data support the notion of definitely higher than normal RV volumes and EF decreased to values associated with RV-PA uncoupling as significant risks of poor outcome after tricuspid valve replacement.
Since pulmonary hypertension is a cause of RV failure with functional tricuspid regurgitation, there is rationale of combining imaging criteria of both the RV and the pulmonary circulation for the purpose of prognostication. A study on 243 patients who underwent trans-catheter tricuspid valve replacement showed an increased rate of clinical deterioration and death during follow-up in 121 of them with pre-operative increase in systolic PAP (sPAP) > 50 mmHg [53••]. The study also showed the independent negative impact on outcome of decreased RV-PA coupling defined by the ratio of tricuspid annular plane systolic excursion (TAPSE) to sPAP < 0.27 mm/mmHg [53••].
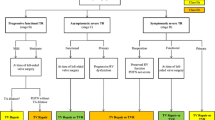
Based on these and other data clinical guidelines give consideration to the repair of severe tricuspid regurgitation in patients with no or minimal symptoms if there is progressive RV dilatation and/or systolic dysfunction [22••]. However the criteria of what constitutes meaningful progression still need to be better defined. The ongoing development of novel trans-catheter tricuspid valve interventions [54] may lead to a change in the landscape of indications for repair in both primary and functional tricuspid regurgitation, as well as redefinition of the roles of RV size and function quantification in guiding timing of therapies. A likely scenario based on currently available data and physiological reasoning is illustrated below in Fig. 7 [55••]. It assumes that patients with a likelihood of pulmonary hypertension will have undergone a confirmatory right heart catheterization as recommended in guidelines on pulmonary hypertension [56]. Also, in keeping with these updated guidelines, a mPAP of 20 mmHg, a PVR of 3 to 6 Wood units and TAPSE/sPAP of 36 mm/mmHg may be more realistic cut-off values for the diagnosis of pulmonary hypertension, RV-PA uncoupling and prediction of post-intervention increase in morbi-mortality [56]. However, large-scale studies are needed for more rigorous definition of cut-off values to predict outcome after tricuspid valve replacement or repair.

Decision-making algorithm based on PVR, mPAP and TAPSE/sPAP for minimized risk of trans-catheter tricuspid valve repair. mPAP, mean pulmonary artery pressure (at right heart catheterization); PVR, pulmonary vascular resistance; RHC, right heart catheterization; sPAP, systolic PAP (at echocardiography); TAPSE, tricuspid annulus plane systolic excursion. Imaging vignettes show TAPSE/sPAP > and < 0.36 mm/mmHg respectively. From ref 55 by D'Alto M, Naeije R. Transcatheter tricuspid valve repair in patients with pulmonary hypertension. Eur Heart J 2020; 41: 2811–281 with permission
Conclusions
Tricuspid regurgitation is a cause of RV volume overload. Over-distension of the RV is associated with irreversible myocardial damage and failure of systolic function adaptation to afterload associated with a risk of poor outcome after tricuspid valve replacement. Current decision-making of this intervention is mainly based on CMR and echocardiographic imaging of the RV with invasive assessment of PVR in case of suspected pulmonary hypertension. Until more robust guidelines established on the basis of large population studies, contra-indications of tricuspid valve replacement include pulmonary hypertension and/or right heart dimensions above the upper limit of normal with RV-PA uncoupling.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Nath J, Foster E, Heidenreich PA. Impact of tricuspid regurgitation on long-term survival. J Am Coll Cardiol. 2004;43:405–9.
Itelman E, Vatury O, Kuperstein R, Ben-Zekry S, Hay I, Fefer P, et al. The association of severe tricuspid regurgitation with poor survival is modified by right ventricular pressure and function: insights from the SHEBAHEART big data. J Am Soc Echocardiogr. 2022;35:1028–36.
•• Sanz J, Sanchez-Quintana D, Bossone E, Bogaard HJ, Naeije R. Anatomy, function, and dysfunction of the right ventricle: JACC State-of-the-Art Review. J Am CollCardiol. 2019;73: 1463–1482. (Important updated guidelines, reviews or recent studies with impact on clinical practice).
•• Sanz J. Volume overload and the right heart. In: Gaine SP, Naeije R and Peacock AJ eds, The right heart, 2nd edition, Springer Nature, Cham, Switzerland. 2021;8:19–136. (Important updated guidelines, reviews or recent studies with impact on clinical practice).
West JB. The role of the fragility of the pulmonary blood-gas barrier in the evolution of the pulmonary circulation. Am J Physiol Regul Integr Comp Physiol. 2013;304:R171-176.
Starr I, Jeffers WA, Meade RH Jr. The absence of conspicuous increments of venous pressure after severe damage to the right ventricle of the dog, with a discussion of the relation between clinical congestive failure and heart disease. Am Heart J. 1943;26:291–301.
Fontan F, Baudet F. Surgical repair of tricuspid atresia. Thorax. 1971;26:240–8.
Gewillig M. The Fontan circulation. Heart. 2005;91:839–46.
Naeije R, Saggar R, Badesch D, Rajagopalan S, Gargani L, Rischard F, et al. Exercise-induced pulmonary hypertension: translating pathophysiological concepts into clinical practice. Chest. 2018;154:10–5.
Paridon SM, Mitchell PD, Colan SD, Williams RV, Blaufox A, Li JS, Margossian R, et al. A cross-sectional study of exercise performance during the first 2 decades of life after the Fontan operation. J Am Coll Cardiol. 2008;52:99–107.
Guyton AC, Lindsey AW, Gilluly JJ. The limits of right ventricular compensation following acute increase in pulmonary circulatory resistance. Circ Res. 1954;2:326–32.
Patterson SW, Piper H, Starling EH. The regulation of the heart beat. J Physiol. 1914;48:465–513.
Rosenblueth A, Alanis J, Lopez E, Rubio R. The adaptation of ventricular muscle to different circulatory conditions. Arch Int Physiol Biochim. 1959;67:358–73.
VonkNoordegraaf A, Chin KM, Haddad F, Hassoun PM, Hemnes AR, Hopkins SR, et al. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J. 2019;53:1801900.
Tello K, Dalmer A, Axmann J, Vanderpool R, Ghofrani HA, Naeije R, et al. Reserve of right ventricular-arterial coupling in the setting of chronic overload. Circ Heart Fail. 2019;12:e005512.
Sanz J, García-Alvarez A, Fernández-Friera L, Nair A, Mirelis JG, Sawit ST, et al. Right ventriculo-arterial coupling in pulmonary hypertension: a magnetic resonance study. Heart. 2012;98:238–43.
Vanderpool RR, Rischard F, Naeije R, Hunter K, Simon MA. Simple functional imaging of the right ventricle in pulmonary hypertension: can right ventricular ejection fraction be improved? Int J Cardiol. 2016;223:93–4.
Badano LP, Addetia K, Pontone G, Torlasco C, Lang RM, Parati G, et al. Advanced imaging of right ventricular anatomy and function. Heart. 2020;106:1469–76.
Sanz J, Conroy J, Narula J. Imaging of the right ventricle. Cardiol Clin. 2012;30:189–203.
Urabe Y, Hamada Y, Spinale FG, Carabello BA, Kent RL, Cooper G 4th, Mann DL. Cardiocyte contractile performance in experimental biventricular volume-overload hypertrophy. Am J Physiol. 1993;264:H1615-1623.
•• Baumgartner H, Bonhoeffer P, De Groot NM, de Haan F, Deanfield JE, Galie N, et al. ESC for the management of gown-up congenital heart disease. Eur Heart J. 2010;31:2915–2957. (Important updated guidelines, reviews or recent studies with impact on clinical practice).
•• Otto CM, Nishimura RA, Bonow RO, Carabello BA, Erwin JP 3d, Gentile F, et al. 2020 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Joint Committee on clinical practice guidelines. J Am CollCardiol. 2021;77:e25-e197. (Important updated guidelines, reviews or recent studies with impact on clinical practice).
Bossers GPL, Hagdorn QAJ, Ploegstra MJ, Borgdorff MAJ, Siljé HW, Beger RMF, et al. Volume load-induced right ventricular dysfunction in animal models: insights in a translational gap in congenital heart disease. Eur J Heart Fail. 2018;20:808–12.
Szabo G, Soos P, Bahrle S, Radovits T, Weigang E, Kekesi V, et al. Adaptation of the right ventricle to an increased afterload in the chronically volume overloaded heart. Ann Thorac Surg. 2006;82:989–95.
Bove T, Vandekerckhove K, Bouchez S, Wouters P, Somers P, Van Nooten G. Role of myocardial hypertrophy on acute and chronic right ventricular performance in relation to chronic volume overload in a porcine model: relevance for the surgical management of tetralogy of Fallot. J Thorac Cardiovasc Surg. 2014;147:1956–65.
Shah AS, Atkins BZ, Hata JA, Tai O, Kypson AP, Lilly RE, et al. Early effects of right ventricular volume overload on ventricular performance and beta-adrenergic signaling. J Thorac Cardiovasc Surg. 2000;120:342–9.
Ersboell M, Vejlstrup N, Nilsson JC, Kjaergaard J, Norman W, Lange T, et al. Percutaneous pulmonary valve replacement after different duration of free pulmonary regurgitation in a porcine model: effects on the right ventricle. Int J Cardiol. 2013;167:2944–51.
Weyman AE, Wann S, Feigenbaum H, Dillon JC. Mechanism of abnormal septal motion in patients with right ventricular volume overload: a cross-sectional echocardiographic study. Circulation. 1976;54:179–86.
Feneley M, Gavaghan T. Paradoxical and pseudoparadoxical interventricular septal motion in patients with right ventricular volume overload. Circulation. 1986;74:230–8.
Chalard A, Sanchez I, Gouton M, Henaine R, Salami FA, Nonet J, et al. Effect of pulmonary valve replacement on left ventricular function in patients with tetralogy of Fallot. Am J Cardiol. 2012;110:1828–35.
Redington AN, Rigby ML, Shinebourne EA, Oldershaw PJ. Changes in the pressure-volume relation of the right ventricle when its loading conditions are modified. Br Heart J. 1990;63:45–9.
Coats L, Khambadkone S, Derrick G, Sridharan S, Schievano S, Mist B, et al. Physiological consequences of percutaneous pulmonary valve implantation: the different behaviour of volume- and pressure-overloaded ventricles. Eur Heart J. 2007;28:1886–93.
Kremer N, Rako Z, Douschan P, Gall H, Ghofrani HA, Grimminger F, et al. Unmasking right ventricular-arterial uncoupling during fluid challenge in pulmonary hypertension. J Heart Lung Transplant. 2022;41:345–55.
Naeije R, Badagliacca R. The overloaded right heart and ventricular interdependence. Cardiovasc Res. 2017;113:1474–85.
Kresoja KP, Rommel KP, Thiele H, Lurz P. Ventricular interaction in a patient with heart failure with preserved ejection fraction and severe tricuspid regurgitation. Circ Heart Fail. 2021;14:e008768.
Lahm T, Douglas IS, Archer SL, Bogaard HJ, Chesler NC, Haddad F, et al. Assessment of right ventricular function in the research setting: knowledge gaps and pathways forward. An official American Thoracic Society research statement. Am J Respir Crit Care Med. 2018;198:e15–43. https://doi.org/10.1164/rccm.201806-1160ST.
Bogaard HJ, Abe K, VonkNoordegraaf A, Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135:794–804.
Ryan JJ, Archer SL. The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ Res. 2014;115:176–88.
Otani H, Kagaya Y, Yamane Y, Chida M, Ito K, Namiuchi S, et al. Long-term right ventricular volume overload increases myocardial fluorodeoxyglucose uptake in the interventricular septum in patients with atrial septal defect. Circulation. 2000;101:1686–92.
Gomez A, Bialostozky D, Zajarias A, Santos E, Palomer A, Martinez MJ, et al. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol. 2001;38:1137–42.
Vogel-Claussen J, Skrok J, Shehata ML, Skrok J, Singh S, Boyce D, et al. Right and left ventricular myocardial perfusion reserves correlate with right ventricular function and pulmonary hemodynamics in patients with pulmonary arterial hypertension. Radiology. 2011;258:119–27.
Zong P, Tune JD, Downey HF. Mechanisms of oxygen demand/supply balance in the right ventricle. Exp Biol Med (Maywood). 2005;230:507–19.
van der Feen DE, Bartelds B, de Boer RA, Berger RMF. Pulmonary arterial hypertension in congenital heart disease: translational opportunities to study the reversibility of pulmonary vascular disease. Eur Heart J. 2017;38:2034–941.
Shiran A, Sagie A. Tricuspid regurgitation in mitral valve disease incidence, prognostic implications, mechanism, and management. J Am Coll Cardiol. 2009;53:401–8.
Topilsky Y, Nkomo VT, Vatury O, Michelena HI, Letourneau T, Suri RM, et al. Clinical outcome of isolated tricuspid regurgitation. JACC Cardiovasc Imaging. 2014;7:1185–94.
Nishimara RA, Otto CM, Bonow RO , Carabello BA, Erwin JP 3d, Guyton RA, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014;63:e57–185.Nath J, Foster E, Heidenreich PA. Impact of tricuspid regurgitation on long-term survival. J Am Coll Cardiol. 2004;43 405–409.
Bartko PE, Arfsten H, Frey MK, Heitzinger G, Pavo N, Cho A, et al. Natural history of functional tricuspid regurgitation: implications of quantitative Doppler assessment. JACC Cardiovasc Imaging. 2019;12:389–97.
Zhan Y, Debs D, Khan MA, Nguyen DT, Graviss EA, Khalaf F, et al. Natural history of functional tricuspid regurgitation quantified by cardiovascular magnetic resonance. J Am Coll Cardiol. 2020;76:1291–301.
Neuhold S, Huelsmann M, Pernicka E, Graf A, Bonderman D, Adlbrecht C, et al. Impact of tricuspid regurgitation on survival in patients with chronic heart failure: unexpected findings of a long-term observational study. Eur Heart J. 2013;34:844–52.
Kim YJ, Kwon DA, Kim HK, Park JS, Hahn S, Kim KH, et al. Determinants of surgical outcome in patients with isolated tricuspid regurgitation. Circulation. 2009;120:1672–8.
Park JB, Kim HK, Jung JH, Klem I, Yoon YE, Lee SP, et al. Prognostic value of cardiac MR imaging for preoperative asessment of patients with severe functional tricuspid regurgitation. Radiology. 2016;280:723–34.
Orban M, Wolff S, Braun D, Stolz L, Higuchi S, Stark K, et al. Right ventricular function in transcatheter edge-to-edge tricuspid valve repair. JACC Cardiovasc Imaging. 2021;14:2477–9.
•• Lurz P, Orban M, Besler C, Braun D, Schlotter F, Noack T, et al. Clinical characteristics, diagnosis, and risk stratification of pulmonary hypertension in severe tricuspid regurgitation and implications for transcatheter tricuspid valve repair. Eur Heart J. 2020;41:2785–95. (Important updated guidelines, reviews or recent studies with impact on clinical practice)
Taramasso M, Benfari G, van der Bijl P, Alessandrini H, Attinger-Toller A, Tiasco L, et al. Transcatheter versus medical treatment of patients with symptomatic severe tricuspid regurgitation. J Am Coll Cardiol. 2019;74:2998–3008.
•• D'Alto M, Naeije R. Transcatheter tricuspid valve repair in patients with pulmonary hypertension. Eur Heart J. 2020;41:2811–281. (Important updated guidelines, reviews or recent studies with impact on clinical practice).
Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;2022(43):3618–731.
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
RN reports relationships with AOP Orphan Pharmaceuticals, Johnson & Johnson, Lung Biotechnology Corporation and United Therapeutics. KT reports relationships with Janssen. MD reports relationships with MSD, Dompe, Ferrer, AOP, and Janssen.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Naeije, R., Tello, K. & D’Alto, M. Tricuspid Regurgitation: Right Ventricular Volume Versus Pressure Load. Curr Heart Fail Rep 20, 208–217 (2023). https://doi.org/10.1007/s11897-023-00599-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-023-00599-w