
Abstract
Purpose of Review
Pancreatic islet cell transplantation is currently the only curative cell therapy for type 1 diabetes mellitus. However, its potential to treat many more patients is limited by several challenges. The emergence of 3D bioprinting technology from recent advances in 3D printing, biomaterials, and cell biology has provided the means to overcome these challenges.
Recent Findings
3D bioprinting allows for the precise fabrication of complex 3D architectures containing spatially distributed cells, biomaterials (bioink), and bioactive factors. Different strategies to capitalize on this ability have been investigated for the 3D bioprinting of pancreatic islets. In particular, with co-axial bioprinting technology, the co-printability of islets with supporting cells such as endothelial progenitor cells and regulatory T cells, which have been shown to accelerate revascularization of islets and improve the outcome of various transplantations, respectively, has been achieved.
Summary
3D bioprinting of islets for generation of an artificial pancreas is a newly emerging field of study with a vast potential to improve islet transplantation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus (DM) is a chronic disorder characterized by hyperglycemia due to failing of glucose metabolism, which causes long-term complications in multiple organs including retinopathy, nephropathy, neuropathy, and vasculopathy [1]. DM is a serious global public health problem, causing significant cost to both the health care system and the global economy. Globally, DM is the eighth leading cause of death causing over 1.5 million deaths directly, and 1.5 million indirectly through hyperglycemia-associated illness such as cardiovascular diseases. In 2014, there were an estimated 422 million adults with diabetes worldwide with the global prevalence of 8.5% and it is predicted to increase to 592 million within 20 years [2].
Type 1 diabetes mellitus (T1DM), also known as juvenile diabetes, accounts for 5–10% of the population with diabetes [3]. Symptoms of T1DM include polyuria, polydipsia, polyphagia, weight loss, blurry vision, and extreme fatigue. T1DM may occur at virtually any age but is most common in children and young adults and occurs as a consequence of an autoimmune destruction of the insulin-producing β cells of the islets of Langerhans in the pancreas, leading to absolute insulin deficiency [1, 2]. The autoimmune destruction is caused by islet-specific T cell response [4] by various autoantibodies such as autoantibodies to insulin [1, 3]. Recent studies suggest T1DM is triggered by environmental factors such as exposure to pathogens or environmental antigens in individuals who are genetically predisposed to diabetes by particular genes such as the HLA genes, which contribute to 50% of the genetic susceptibility to T1DM [2, 3].
Currently, patients with T1DM are treated with daily exogenous insulin administration [5, 6]. However, despite advances in medicine, there has not yet been a development of an insulin therapy that can mimic the physiological rhythms or a mechanical replacement for pancreatic β cells. An intensive monitoring of blood glucose level accompanied by exogenous insulin therapy via insulin injection or pump represents the current state of treatments for T1DM. Although these treatments are able to delay the progression of diabetic complications including neuropathy and retinopathy, it is not sufficient to prevent these complications [7]. The replacement of β cell function through whole pancreas transplantation is presently the only permanent alternative for re-establishing endogenous insulin secretion in patients with T1DM [8].
Current Approaches for β Cell Replacement
Pancreas transplantation is reserved and performed in patients with T1DM and advanced or end-stage renal disease. As a result, over three-quarters of the whole pancreas is transplanted in conjunction with kidney transplantation as either simultaneous kidney-pancreas transplantation or alternatively pancreas after kidney transplantation [9]. Furthermore, the surgical procedure is associated with significant mortality risk, accompanied with clinically significant complications such as pancreatitis, bleeding, re-occurrence of autoimmunity, and rejection post-transplantation, which motivates the urgent need for the search of an alternative therapy [10].
A logical alternative to whole organ transplantation is to transplant the cells that have been destroyed. Pancreatic islet transplantation is a minimally invasive approach where purified allogeneic donor islets, isolated from deceased organ donor pancreata, are currently percutaneously infused into recipient liver through the portal vein [11,12,13]. This procedure has lower risk compared with pancreas transplantation, as major surgery is not required and a differing immunosuppression regimen is employed. A cellular approach was first tried unsuccessfully in man in 1894 using fragmented sheep pancreas in a subject with diabetes [12, 14]. The successful application of islet transplantation as a treatment for diabetes was not realized for many decades until reversal of diabetes was initially observed in rodents and in a patient with chronic pancreatitis who underwent pancreatectomy followed by islet autotransplantation [15,16,17]. Following these findings, intensive research has been conducted in the field of islet transplantation. In 2000, the Edmonton immunosuppression protocol, which utilized a corticosteroid-free immunosuppression regimen and multiple islet infusions from different donors, was established [18]. An insulin independence rate of 100% was achieved in seven patients following 1 year of islet transplantation and partial graft function was observed in most of the seven patients after 5 years [18, 19] which represented a significant improvement from the success rate of 10% prior to the protocol [12, 20, 21]. Critically long-term insulin independence has been difficult to achieve, and most patients require at least two infusions to achieve insulin independence [19]. Islet transplantation has been adopted as a treatment option for T1DM in a number of countries and has proved an attractive method of β cell replacement [21].
Despite the significant progress in islet transplantation procedures, numerous obstacles remain that currently limit its clinical application [8, 22]. The current clinical standard of care involves the infusion of islets into the patient liver via the portal vein where islets encounter a sub-optimal non-pancreatic environment: high glucose concentration, lower oxygen tension, and higher level of toxins [23]. Moreover, infusion of islets via the hepatic portal vein triggers an innate immune reaction upon contact with blood, known as the instant blood-mediated inflammatory reaction [24]. The hypoxic islets secrete chemokines and express tissue factors, which activates a thrombotic reaction [25]. Platelets are attracted to the islet surface, recruiting leukocytes and macrophages to infiltrate and destroy the islet cells [24, 26]. Together, these factors kill up to 70% of transplanted islets in the first 48 h [27, 28] and consequentially islets from up to three donor pancreata are required for clinical benefit, limiting the availability of the transplantation. Additionally, the obligatory use of immunosuppressive regimen is another major challenge of islet transplantation. Immunosuppressive drugs used for islet transplantation are associated with many side effects including risks of infection, higher rate of malignancy, β cell toxicity, and organ toxicity, significantly decreasing the individual’s quality of life [20].
Alternative Transplantation Site
Many studies in recent years have explored alternative sites for islet transplantation with the following key characteristics: (a) sufficient space for islet engraftment; (b) close proximity to vascular network to provide optimal oxygen tension, sensing, and release of insulin; (c) allow real-time communication between cellular graft and the circulation; and (d) offer minimal inflammatory potential to support long-term graft survival. A few sites with immunological privileges such as the testis or thymus have been tested in small animals; however, to date, they remained clinically irrelevant due to limited space for islet engraftment [29, 30]. Among many sites explored, the skin site received attention as it offers a readily accessible site via a minimally invasive surgical procedure. The only drawback is that unlike the liver or kidney capsule, dermal poor vascularization limits the integration and functionality of engrafted islets [31]. The pancreas, the native home of islets, has also been explored as a site of islet transplantation. However, it is not considered for a transplantation site due to the metabolic complications such as pancreatitis (potentially induced after embolization) and limited vascular supply. At this point in time, for clinical islet transplantation, intra-portal infusion remains the gold standard [32].
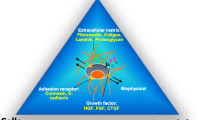
As no suitable alternative transplantation site in the human body has been found, one option to explore is the fabrication of an artificial transplantation site. The recent advancements in bio-engineering technology now enable constructing of such sites. Hydrogels are a multi-component system comprised of a tri-dimensional network of polymer chains with absorbed water filling the space between the macromolecules, within which various biomaterials may be incorporated to mimic tissue-like properties. To date, various types of naturally derived polymers (e.g., alginate, collagen, gelatin, fibrin, and fibronectin) and synthetic polymers (e.g., poly (lactic-co-glycolic acid), polysulfone, poly (lactic acid), poly (vinyl alcohol)) have been evaluated [33]. Among all, alginate-based hydrogels have been the most extensively investigated polymers for their utility with pancreatic islets to treat T1DM. Alginate is a naturally occurring anionic polymer typically obtained from brown seaweed. It is bio-inert, and naturally hydrophilic, thus allowing covalent functionalization via interaction with extracellular matrix proteins, peptides, and growth factors [34, 35].
The main approach of incorporating such technology in islet transplantation is via islet encapsulation. Pancreatic islets are embedded within a hydrogel, and this “mini organ” holds pores which allow bidirectional diffusion of small molecules such as insulin (~ 6 kDa), nutrients, and glucose, and at the same time, protects islets from immune attack by restricting the access of immune cells or antibodies (~ 150–900 kDa) [36]. Numerous small animal models have demonstrated that islet encapsulation is able to improve glucose homeostasis for a short term, but no permanent restoration of euglycemia was observed [37,38,39,40,41]. Several challenges are suggested to limit this biomedical approach to advance into clinical settings. One challenge arises as physical irregularities from fabricating hydrogel result in an incomplete coverage of the islets within the capsules. This may trigger a pericapsular fibrotic overgrowth (PFO) which blocks the diffusion of nutrients and oxygen, resulting in islet necrosis [42, 43]. Even the successfully encapsulated islets suffer from hypoxia due to the restricting hydrogel permeability and increased distance from the surrounding blood vessels reduces the availability of oxygen by diffusion. This introduces a challenge in scaling up into a large animal model or clinically relevant dose of islets. Moreover, encapsulation prevents immediate revascularization post-transplantation, subjecting the islets to further hypoxic stress. As hypoxia hampers the function and responsiveness of islets to glucose, even a larger number of islets are needed to restore normoglycemia.
3D Bioprinting
One approach to address hypoxia involves “seeding” of islets onto degradable 3D scaffold structures [44]. Scaffolds are made of similar biopolymers to mimic the pancreatic microenvironment. The construct provides increased surface area to volume ratio compared with the hydrogel capsules, and even allows for vascular ingrowth, thereby providing increased oxygen and nutrient supply [45]. Upon slow degradation of the scaffold, the extracellular matrix proteins are deposited by surrounding tissues and engrafted islets, gradually re-building the suitable environment required for islet survival [46]. Even though immune isolation is not achieved through this method, the scaffolds can prevent direct contact of embedded islets to circulating immune cells to reduce the inflammatory response until the scaffolds eventually degrades [24]. The efficacy of such device has been demonstrated in animal studies [47,48,49]. Moreover, utilization of 3D bioprinting technology with higher accuracy could provide highly controlled seeding of islets, thereby minimizing the onset of PFO arising with conventional techniques.
The concept of 3D printing was first introduced in 1986 by Charles W. Hull and has become increasingly prominent over the past decades [50]. 3D printing technology allows printing of a 3D structure, typically through stacking successive thin layers in a layer-by-layer fashion. Advances in engineering technology have now opened up the possibility of using 3D printing to “print” spatially controlled biomaterial structures with embedded bioactive factors and cells into a functional tissue construct [51]. Such automated printing allows for the precise control of architecture, pore interconnectivity, and a high degree of reproducibility necessary for commercial clinical application and regulation. Furthermore, bioprinting allows the deposition of a wide array of cell types and bioactive factors in a precise order to simulate native tissue environment and support cell survival [52,53,54].
The current varieties of 3D bioprinting techniques include extrusion printing, inkjet printing, laser-assisted bioprinting, and stereolithography bioprinting [55]. Among these, extrusion bioprinting has been most extensively investigated for the generation of artificial tissue constructs such as cartilages [56, 57], liver [58], and neural tissues [59], with its ability to print various biomaterials at high cell density [60]. Extrusion bioprinting has been utilized for the generation of artificial pancreata. A rat β cell line, mouse and human islets were successfully printed into a predefined 3D scaffold using alginate-based bioinks and the subsequent cell viability and morphology were found to be unaffected [61••]. Furthermore, rat islets were printed into macroporous 3D constructs using an alginate/methylcellulose bioink. These printed rat islets retained their viability, morphology, and function, for up to 7 days in culture [62••].
Beyond the modification of the bioinks to support islet cells, bioprinting also enables the co-transplantation of islets with supporting cells that could enhance islet survival [63]. Recently, our group developed a 3D bioprinter equipped with a co-axial extruder nozzle and two separate ink chambers [64••]. Different bioinks tailored with cell type–specific bioactive molecules can be utilized in each chamber, allowing co-printing of islets with supporting cells. These geometries have the advantage that more delicate components can be strategically placed within the core with a surrounding protective layer, referred to as the shell. The use of a co-axial structure has been shown to significantly improve islet encapsulation by minimizing material volume per islet and reducing the risk of PFO [42]. Co-axially printed mouse islets and endothelial progenitor cells (EPCs) demonstrated high viability using recently formulated alginate-gelatin methacryloyl bioink [64••]. Together, co-axial 3D bioprinting has the potential to enable the embedding of clinically relevant doses of islets with support for islet survival, including supporting cells and bioactive factors.
Supporting Cells
Endothelial Progenitor Cells
The islet of Langerhans is densely vascularized with fenestrated endothelium, and this feature is crucial for β cells to readily sense the blood glucose and secrete insulin into the systemic circulation [65]. Endothelial cells are also crucial for promoting islet function through upregulating insulin transcription via either cell-to-cell contact mechanism or secretion of humoral factors [66]. However, islets are removed from the vasculature during isolation and remain avascular for up to several days post-transplantation [67]. As native islets are physiologically adapted to receiving a rich supply of oxygen (the islet mass in the pancreas comprises 1% of the cells but receives 10% of the blood supply), prolonged hypoxia is detrimental to islet viability and function. The hypoxia induced by isolation triggers activation of NF-kB and inducible nitric oxide synthase, leading to the production of cytotoxic nitric oxide [68, 69]. The consequence of free radical production is decreased insulin production, macrophage cell infiltration, and islet apoptosis. Moreover, islets exposed to hypoxia pre-transplantation display dysregulated glucose responsiveness and elevated basal insulin secretion, due to the upregulation of hypoxic response genes which can continue post-transplantation, resulting in poor glycemic control [70]. Normally, the revascularization occurs 2–4 days post-transplantation and takes over 2 weeks to complete [71]. The eventual vascular density and oxygen tension in transplanted engraftments are less than 50% as compared with the native pancreatic islets [72]. This highlights the crucial need for a mechanism to improve revascularization to restore full function of engrafted islets.
One approach to achieving such acceleration is to directly use pro-angiogenic cells such as EPCs. EPCs are a heterogeneous group of stem cells derived from the bone marrow that are recruited to the site of hypoxia. EPCs promote vascularization by recruiting and differentiating other cell types, or by differentiating themselves into endothelial cells, the building blocks of the endothelial lining of blood vessels [73, 74]. Furthermore, the crosstalk between intra-islet endothelial cells and islets enhances expression and secretion of insulin and islet survival [75]. These properties of EPCs were shown to be beneficial when EPCs were co-transplanted with islets in several studies. Co-transplantation of human umbilical cord–derived EPCs with porcine islets in diabetic nude mice significantly accelerated the revascularization in the first 2 weeks, compared with islet transplantation alone, and increased expression of pro-angiogenic factor, vascular endothelial growth factor-A (VEGF-A), in islets [76]. Co-transplantation of rat bone marrow–derived EPCs with rat islets in diabetic rats demonstrated long-lasting normoglycemia [77]. Moreover, co-transplantation of mouse bone marrow–derived EPCs with mouse islets in diabetic mice improved the cure rate and glucose control with higher final β cell mass [78•].
Regulatory T Cells
Upon transplantation, recipient T cells recognize alloantigens of the allograft, which activate recipient T cells to induce inflammation at the site of the allograft, as part of the alloresponse [79]. Alloresponse results in acute graft rejection as well as chronic graft dysfunction, and as such the use of immunosuppressive drugs is necessitated to prevent the commencement of alloresponse. However, as aforementioned, immunosuppressants are associated with deleterious side effects; there is a need for novel immunotherapies to achieve immunosuppression-free transplantation. Regulatory T cells (Tregs) are a promising candidate for this.
Tregs are a sub-population of T cells that specialize in immune regulation and suppression. Tregs are defined as CD4+ FOXP3+ CD25hi CD127− T cells with two main types of Tregs. Natural Tregs (nTregs) arise from highly self-reactive T cells in the thymus during T cell development and establishment of central tolerance [80]. nTregs are characterized by complete demethylation of the FOXP3 promoter, which results in stable expression of FOXP3 and a suppressive phenotype [81]. On the other hand, peripheral or induced Tregs are generated from naïve CD4+ T cells in the periphery or in vitro upon stimulation with transforming growth factor beta (TGF-β) and retinoic acid. The FOXP3 promoter of induced Tregs is only partially demethylated leading to their functional instability. Treg-mediated suppression is the main mechanism of peripheral tolerance. Tregs play many roles in the immune system including downregulation of immune responses once pathogens have been cleared, maintaining tolerance to self, gut microbiota, new chemicals, environmental and food antigens, and the fetus in cases of pregnancy. Treg-mediated suppression targets a broad spectrum of immune cells such as other T cell subsets, B cells, antigen-presenting cells, and natural killer cells [82]. Suppression is achieved via several circumstance-dependent mechanisms, including inhibitory cytokines, cytolysis, metabolic disruption, and modulation of dendritic cells [83].
Since the first organ transplantation clinical trials, spontaneous graft tolerance (the acceptance of allograft without immunosuppressive regimen) has been observed, mostly in liver transplant recipients [84,85,86]. The exact factors underlying this phenomenon have not yet been fully identified; however, it has been shown that Tregs play a major role in spontaneous graft tolerance in mice liver allograft models [87, 88] and there is elevation of Treg proportion in spontaneously tolerant patients [89]. Thus, various approaches to utilize their natural abilities have been extensively investigated. In particular, adoptive transfer of ex vivo expanded Tregs has a number of advantages including greater control over the expansion/generation of Tregs, the possibility of functional and phenotypical analysis prior to delivery, and finer control of dosage and delivery time [90, 91]. This has proved promising in murine islet transplantation models [92, 93] and in clinical trials for kidney [94•] and liver [95•] transplantation.
Bioactive Factors
Insulin-Like Growth Factor-2
Insulin-like growth factor-2 is a potent growth factor highly expressed in islets during fetal development with much less activity in post-natal life [96, 97]. The fetal overexpression of IGF-II can enlarge individual organs, and the whole body size of a new born murine, in a dose-dependent manner [98]. Notably, the expression pattern of IGF-II in pre-natal period coincides with the pancreatic mass growth, suggesting its role in proliferation or survival of β cells [99, 100]. In vitro, IGF-II showed increased DNA replication in β cell lines, and IGF-II survival effect on transplanted islets has been observed in animal models [101,102,103]. Together, it is suggested that IGF-II plays a dual beneficial role on β cells as a survival and mitogenic factor. In addition, IGF-II pre-incubation or viral transfection-induced overexpression of IGF-II can effectively improve islet survival against cytokine exposure and islet engraftment [104]. As such, incorporation of IGF-II could protect islets from hypoxia and cytokine-driven cell deaths until revascularization is established.
Vascular Endothelial Growth Factor
VEGF is a family of pro-angiogenic proteins which includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, VEGF-F, and PIGF (placental growth factor) in human. Among these, VEGF-A plays a crucial role in vasculogenesis during embryogenesis as well as angiogenesis in post-natal life [105]. Furthermore, VEGF-A is required for formation of a dense network of fenestrated blood vessels around islets [106]. These properties give VEGF-A the therapeutic potential to induce neovascularization in islet transplantation. VEGF-A containing alginate scaffolds were demonstrated to pre-vascularize murine intramuscular space prior to islet transplantation and to improve islet survival after transplantation [107]. In addition, 3D ring-shaped polycaprolactone scaffolds functionalized with VEGF-A were able to induce vascularization and improve function of encapsulated islets embedded in the polycaprolactone scaffolds, compared with free-floating islets [108•]. Thus, incorporation of VEGF-A in conjunction with EPCs could accelerate revascularization of the islets.
Interleukin-2
IL-2 is a T cell stimulatory cytokine, largely produced by CD4+ T helper cells. IL-2 signaling is crucial for activation and clonal expansion T cells [109]. While CD4+ T helper cells can produce IL-2 for autocrine signaling upon T cell receptor stimulation, Treg cells cannot and thus are reliant on IL-2 produced by other cells [110, 111]. IL-2 is crucial for Treg function as well as their survival. Tregs highly express CD25, the α-chain of the high-affinity IL-2 receptor complex [112] and the interaction of IL-2 and CD25 induces high expression of FOXP3 thereby reinforcing Treg phenotype and function [113, 114]. Moreover, with high expression of CD25, Tregs can respond to low concentrations of IL-2 and bind to IL-2 with high affinity [115], which can lead to sequestration of IL-2 from effector T cells, depriving them of survival signal [83]. Administration of exogenous IL-2 in autoimmunity and organ transplantation has been investigated to augment Treg numbers and function [116]. Particularly, low-dose IL-2 therapy has shown promising results in hematopoietic stem cell transplantation graft-versus-host disease, selectively increasing Treg numbers [117,118,119,120]. Thus, incorporation of IL-2 may enhance survival and function of printed Tregs and create a Treg-rich microenvironment around the 3D bioprinted scaffold.
Conclusion
Pancreatic islet transplantation is a promising curative cell therapy for T1DM. The field is limited by human cadaveric islet cell sources at present, but newer cell sources such as embryonic stem cell or xenogeneic cell sources are in the pipeline. The current procedure of islet infusion into liver may not be optimal for these newer cell sources, and so alternative transplant strategies such as 3D bioprinting may provide new strategies. Modifications of the bioinks with local immunosuppression and bioactive factors to support the cells are new directions for the field. The recent 3D bioprinting technology, especially with the development of the co-axial bioprinter, thus has potential to change the current pancreatic islet transplantation paradigm. The possibility of co-printing islets with supporting cells and bioactive factors potentiates direct improvement of engraftment condition, and thus survival and function of transplanted islets. Incorporation of EPCs may accelerate revascularization of islets to support their function, and the incorporation of Tregs could provide localized immune protection to islets. Bioactive factors such IGF-II, VEGF, and IL-2 could enhance the survival and function of printed cells to maximize the efficacy of the graft. Together, extra-hepatic islet transplantation without the use of immunosuppression might be clinically achieved by utilizing 3D bioprinting technology.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Gregory JM, Moore DJ. Type 1 diabetes mellitus. Pediatr Rev 2013. https://doi.org/10.1016/B978-1-4377-1604-7.00561-3.
Forouhi NG, Wareham NJ. Epidemiology of diabetes. Medicine (Baltimore). 2014;42:698–702. https://doi.org/10.1383/medc.2006.34.2.57.
Diabetes DOF. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2013;36:67–74. https://doi.org/10.2337/dc13-S067.
Pathiraja V, Kuehlich JP, Campbell PD, Krishnamurthy B, Loudovaris T, Coates PTH, et al. Proinsulin-specific, HLA-DQ8, and HLA-DQ8-transdimer-restricted CD4+ T cells infiltrate islets in type 1 diabetes. Diabetes. 2015;64:172–82. https://doi.org/10.2337/db14-0858.
Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32:1121–33. https://doi.org/10.1038/nbt.3033.
Prabakar KR, Domínguez-Bendala J, Damaris Molano R, Pileggi A, Villate S, Ricordi C, et al. Generation of glucose-responsive, insulin-producing cells from human umbilical cord blood-derived mesenchymal stem cells. Cell Transplant. 2012;21:1321–39. https://doi.org/10.3727/096368911X612530.
Agarwal A, Brayman KL. Update on islet cell transplantation for type 1 diabetes. Semin Interv Radiol. 2012;29:90–8. https://doi.org/10.1055/s-0032-1312569.
Robertson RP, Davis C, Larsen J, Stratta R, Sutherland DER, American Diabetes Association. Pancreas and islet transplantation in type 1 diabetes. Diabetes Care. 2006;29:935. https://doi.org/10.2337/diacare.29.04.06.dc06-9908.
Chan C, Chim TM, Leung K, Tong C, Wong T, Leung GK. Simultaneous pancreas and kidney transplantation as the standard surgical treatment for diabetes mellitus patients with end-stage renal disease. Hong Kong Med J. 2016;22(1):62–9. https://doi.org/10.12809/hkmj154613.
Troppmann C. Complications after pancreas transplantation. Curr Opin Organ Transpl. 2010;15:112–8. https://doi.org/10.1097/MOT.0b013e3283355349.
Vardanyan M, Parkin E, Gruessner C, Rilo HLR. Pancreas vs. islet transplantation: a call on the future. Curr Opin Organ Transplant. 2010;15:124–30. https://doi.org/10.1097/MOT.0b013e32833553f8.
Robertson RP. Islet transplantation as a treatment for diabetes — a work in progress. N Engl J Med. 2004;350:694–705. https://doi.org/10.1056/NEJMra032425.
Connell PJO, Holmes-Walker DJ, Goodman D, Hawthorne WJ, Loudovaris T, Gunton JE, et al. Multicenter Australian trial of islet transplantation: improving accessibility and outcomes. Am J Transplant. 2013;13:1850–8. https://doi.org/10.1002/ajt.12250.
Ichii H, Ricordi C. Current status of islet cell transplantation. J Hepato-Biliary-Pancreat Surg. 2013;16:101–12. https://doi.org/10.1007/s00534-008-0021-2.Current.
Lacy PE, Walker MM, Fink CJ, Louis S. Perifusion of isolated rat islets in vitro participation of the microtubular system in the biphasic release of insulin. Diabetes. 1972;21:987–98. https://doi.org/10.2337/diab.21.10.987.
Najarian JS, Sutherland DER, Baumgartner D, Burke B, Rynasiewicz JJ, Matas AJ, et al. Total or near total pancreatectomy and islet autotransplantation for treatment of chronic pancreatitis.pdf. Ann Surg. 1980;192:526–42.
Robertson RP, Lanz KJ, Sutherland DER, Kendall DM. Prevention of diabetes for up to 13 years by autoislet transplantation after pancreatectomy for chronic pancreatitis. Diabetes. 2001;50:48–50. https://doi.org/10.2337/diabetes.50.1.47.
Shapiro AMJ, Lakey JRT, Ryan EA, Korbutt GS, Toth E, Warnock GL, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocoricoid-free imunosuppressive regimen. N Engl J Med. 2000;343:230–8. https://doi.org/10.1056/NEJM200007273430401.
Ryan EA, Paty BW, Senior PA, Bigam D, Alfadhli E, Kneteman NM, et al. Five-year follow-up after clinical islet transplantation. Diabetes. 2005;54:2060–9.
Leitão CB, Cure P, Tharvanji T, Baidal DA, Alejandro R. Current challenges in islet transplantation. Curr Diab Rep. 2008;8:324–31. https://doi.org/10.1007/s11892-008-0057-3.
Rheinheimer J, Bauer AC, Silveiro SP, Estivalet AAF, Bouças AP, Rosa AR, et al. Human pancreatic islet transplantation: an update and description of the establishment of a pancreatic islet isolation laboratory. Arch Endocrinol Metab. 2015;59:161–70. https://doi.org/10.1590/2359-3997000000030.
Lacy PE, Kostianovsky M. Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes. 1967;16:35–9. https://doi.org/10.2337/diab.16.1.35.
Carlsson PO, Palm F, Andersson A, Liss P. Markedly decreased oxygen tension in transplanted rat pancreatic islets irrespective of the implantation site. Diabetes. 2001;50:489–95. https://doi.org/10.2337/diabetes.50.3.489.
Bennet W, Sundberg B, Groth CG, Brendel MD, Brandhorst D, Brandhorst H, et al. Incompatibility between human blood and isolated islets of Langerhans: a finding with implications for clinical intraportal islet transplantation? Diabetes. 1999;48:1907–14. https://doi.org/10.2337/diabetes.48.10.1907.
Moberg L, Johansson H, Lukinius A, Berne C, Foss A, Källen R, et al. Production of tissue factor by pancreatic islet cells as a trigger of detrimental thrombotic reactions in clinical islet transplantation. Lancet. 2002;360:2039–45. https://doi.org/10.1016/S0140-6736(02)12020-4.
Van Der Windt DJ, Bottino R, Casu A, Campanile N, Cooper DKC. Rapid loss of intraportally transplanted islets: an overview of pathophysiology and preventive strategies. Xenotransplantation. 2007;14:288–97. https://doi.org/10.1111/j.1399-3089.2007.00419.x.
Cantarelli E, Piemonti L. Alternative transplantation sites for pancreatic islet grafts. Curr Diab Rep. 2011;11:364–74. https://doi.org/10.1007/s11892-011-0216-9.
Bennet W, Groth CG, Larsson R, Nilsson B, Korsgren O. Isolated human islets trigger an instant blood mediated inflammatory reaction: implications for intraportal islet transplantation as a treatment for patients with type 1 diabetes. Ups J Med Sci. 2000;105:125–33. https://doi.org/10.1517/03009734000000059.
Margolis RN, Holup JJ, Selawry HP. Effects of intratesticular islet transplantation on hepatic glycogen metabolism in the rat. Diabetes Res Clin Pract. 1986;2:291–9. https://doi.org/10.1016/S0168-8227(86)80006-7.
Ar’Rajab A, Dawidson IJ, Harris RB, Sentementes JT. Immune privilege of the testis for islet xenotransplantation (rat to mouse). Cell Transplant. 2017;3:493–8. https://doi.org/10.1177/096368979400300606.
Kemp CB, Knight MJ, Scharp DW, Ballinger WF, Lacy PE. Effect of transplantation site on the results of pancreatic islet isografts in diabetic rats. Diabetologia. 1973;9:486–91. https://doi.org/10.1007/BF00461694.
Melzi R, Sanvito F, Mercalli A, Andralojc K, Bonifacio E, Piemonti L. Intrahepatic islet transplant in the mouse: functional and morphological characterization. Cell Transplant. 2008;17:1361–70. https://doi.org/10.3727/096368908787648146.
Mao G, Chen G, Bai H, Song T, Wang Y. The reversal of hyperglycaemia in diabetic mice using PLGA scaffolds seeded with islet-like cells derived from human embryonic stem cells. Biomaterials. 2009;30:1706–14. https://doi.org/10.1016/j.biomaterials.2008.12.030.
De Vos P, Hamel AF, Tatarkiewicz K. Considerations for successful transplantation of encapsulated pancreatic islets. Diabetologia. 2002;45:159–73. https://doi.org/10.1007/s00125-001-0729-x.
Mallett AG, Korbutt GS. Alginate modification improves long-term survival and function of transplanted encapsulated islets. Tissue Eng Part A. 2009;15:1301–9. https://doi.org/10.1089/ten.tea.2008.0118.
Desai T, Shea LD. Advances in islet encapsulation technologies. Nat Rev Drug Discov. 2017;16:338–50. https://doi.org/10.1038/nrd.2016.232.
Vériter S, Mergen J, Goebbels R-M, Aouassar N, Grégoire C, Jordan B, et al. In vivo selection of biocompatible alginates for islet encapsulation and subcutaneous transplantation. Tissue Eng Part A. 2010;16:1503–13. https://doi.org/10.1089/ten.TEA.2009.0286.
Lum ZP, Tai IT, Krestow M, Norton J, Vacek I, Sun AM. Prolonged reversal of diabetic state in NOD mice by xenografts of microencapsulated rat islets. Diabetes. 1991;40:1511–6. https://doi.org/10.2337/diab.40.11.1511.
Cui W, Barr G, Faucher KM, Sun X-L, Safley SA, Weber CJ, et al. A membrane-mimetic barrier for islet encapsulation. Transplant Proc. 2004;36:1206–8. https://doi.org/10.1016/j.transproceed.2004.04.059.
Lamb M, Storrs R, Li S, Liang O, Laugenour K, Dorian R, et al. Function and viability of human islets encapsulated in alginate sheets: in vitro and in vivo culture. Transplant Proc. 2011;43:3265–6. https://doi.org/10.1016/j.transproceed.2011.10.028.
Zhi ZL, Kerby A, King AJF, Jones PM, Pickup JC. Nano-scale encapsulation enhances allograft survival and function of islets transplanted in a mouse model of diabetes. Diabetologia. 2012;55:1081–90. https://doi.org/10.1007/s00125-011-2431-y.
Ma M, Chiu A, Sahay G, Doloff JC, Dholakia N, Thakrar R, et al. Core-shell hydrogel microcapsules for improved islets encapsulation. Adv Healthc Mater. 2013;2:667–72. https://doi.org/10.1002/adhm.201200341.
Hobbs HA, Kendall WF, Darrabie M, Opara EC. Prevention of morphological changes in alginate microcapsules for islet xenotransplantation. J Investig Med. 2001;49:572–5. https://doi.org/10.2310/6650.2001.33722.
De Vos P, Hillebrands JL, De Haan BJ, Strubbe JH, Van Schilfgaarde R. Efficacy of a prevascularized expanded polytetrafluoroethylene solid support system as a transplantation site for pancreatic islets. Transplantation. 1997;63:824–30. https://doi.org/10.1097/00007890-199703270-00006.
Dufour JM, Rajotte RV, Zimmerman M, Rezania A, Kin T, Dixon DE, et al. Development of an ectopic site for islet transplantation, using biodegradable scaffolds. Tissue Eng. 2005;11:1323–31. https://doi.org/10.1089/ten.2005.11.1323.
Wang RN, Rosenberg L. Maintenance of beta-cell function and survival following islet isolation requires re-establishment of the islet-matrix relationship. J Endocrinol. 1999;163:181–90. https://doi.org/10.1677/joe.0.1630181.
Gibly RF, Zhang X, Graham ML, Hering BJ, Kaufman DB, Lowe WL, et al. Extrahepatic islet transplantation with microporous polymer scaffolds in syngeneic mouse and allogeneic porcine models. Biomaterials. 2011;32:9677–84. https://doi.org/10.1016/j.biomaterials.2011.08.084.
Berman DM, O’Neil JJ, Coffey LCK, Chaffanjon PCJ, Kenyon NM, Ruiz P, et al. Long-term survival of nonhuman primate islets implanted in an omental pouch on a biodegradable scaffold. Am J Transplant. 2009;9:91–104. https://doi.org/10.1111/j.1600-6143.2008.02489.x.
Blomeier H, Zhang X, Rives C, Brissova M, Hughes E, Baker M, et al. Polymer scaffolds as synthetic microenvironments for extrahepatic islet transplantation. Transplantation. 2006;82:452–9. https://doi.org/10.1097/01.tp.0000231708.19937.21.
Murphy SV, Atala A. 3D bioprinting of tissues and organs. Nat Biotechnol. 2014;32:773–85. https://doi.org/10.1038/nbt.2958.
Schubert C, van Langeveld MC, Donoso LA. Innovations in 3D printing: a 3D overview from optics to organs. Br J Ophthalmol. 2014;98:159–61. https://doi.org/10.1136/bjophthalmol-2013-304446.
Ozbolat IT, Chen H, Yu Y. Development of ‘multi-arm bioprinter’ for hybrid biofabrication of tissue engineering constructs. Robot Comput Integr Manuf. 2014;30:295–304. https://doi.org/10.1016/j.rcim.2013.10.005.
Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA, Lewis JA. 3D bioprinting of vascularized, heterogeneous cell-laden tissue constructs. Adv Mater. 2014;26:3124–30. https://doi.org/10.1002/adma.201305506.
Poldervaart MT, Gremmels H, van Deventer K, Fledderus JO, Oner FC, Verhaar MC, et al. Prolonged presence of VEGF promotes vascularization in 3D bioprinted scaffolds with defined architecture. J Control Release. 2014;184:58–66. https://doi.org/10.1016/j.jconrel.2014.04.007.
Yue Z, Liu X, Coates PT, Wallace GG. Advances in printing biomaterials and living cells. Curr Opin Organ Transplant. 2016;21:467–75. https://doi.org/10.1097/MOT.0000000000000346.
Di Bella C, Duchi S, O’Connell CD, Blanchard R, Augustine C, Yue Z, et al. In situ handheld three-dimensional bioprinting for cartilage regeneration. J Tissue Eng Regen Med. 2018;12:611–21. https://doi.org/10.1002/term.2476.
Kesti M, Eberhardt C, Pagliccia G, Kenkel D, Grande D, Boss A, et al. Bioprinting complex cartilaginous structures with clinically compliant biomaterials. Adv Funct Mater. 2015;25:7406–17. https://doi.org/10.1002/adfm.201503423.
Lee JW, Choi Y-J, Yong W-J, Pati F, Shim J-H, Kang KS, et al. Development of a 3D cell printed construct considering angiogenesis for liver tissue engineering. Biofabrication. 2016;8:015007. https://doi.org/10.1088/1758-5090/7/2/025009.
Gu Q, Tomaskovic-crook E, Lozano R, Chen Y, Kapsa RM, Zhou Q, et al. Functional 3D neural mini-tissues from printed gel-based bioink and human neural stem cells. Adv Healthc Mater. 2016;5:1429–38. https://doi.org/10.1002/adhm.201600095.
Derakhshanfar S, Mbeleck R, Xu K, Zhang X, Zhong W, Xing M. 3D bioprinting for biomedical devices and tissue engineering: a review of recent trends and advances. Bioact Mater Elsevier Ltd. 2018;3:144–56. https://doi.org/10.1016/j.bioactmat.2017.11.008.
•• Marchioli G, Van Gurp L, Van Krieken PP, Stamatialis D, Engelse M, Van Blitterswijk CA, et al. Fabrication of three-dimensional bioplotted hydrogel scaffolds for islets of Langerhans transplantation. Biofabrication. 2015;7:025009. https://doi.org/10.1088/1758-5090/7/2/025009 3D bioprinting technology was used to print pancreatic islets using alginate-based bioinks. 3D bioprinted human islets were highly viable and morphologically sound.
•• Duin S, Schütz K, Ahlfeld T, Lehmann S, Lode A, Ludwig B, et al. 3D bioprinting of functional islets of Langerhans in an alginate/methylcellulose hydrogel blend. Adv Healthc Mater. 2019;1801631:1–14. https://doi.org/10.1002/adhm.201801631 3D bioprinting technology was used to print rat islets using an alginate/methylcellulose bioink with minimal impact on islet viability , morphology, and function.
Penko D, Mohanasundaram D, Sen S, Mee C, Bonder CS, Coates PTH, et al. Incorporation of endothelial progenitor cells into mosaic pseudoislets. Islets. 2011;3:73–9. https://doi.org/10.4161/isl.3.3.15392.
•• Liu X, Carter SSD, Renes MJ, Kim J, Rojas-Canales DM, Penko D, et al. Development of a coaxial 3D printing platform for biofabrication of implantable islet-containing constructs. Adv Healthc Mater. 2019;8(7):e1801181. https://doi.org/10.1002/adhm.201801181 A co-axial bioprinter was developed in-house and alginate-gelatin methacryloyl bioink was formulated, for co-printing of islets with supporting cells such as endothelial progenitor cells and regulatory T cells. Co-printed mouse islets and endothelial progenitor cells were highly viable.
Lifson N, Lassa CV, Dixit PK. Relation between blood flow and morphology in islet organ of rat pancreas. Am J Phys. 1985;249:E43–8. https://doi.org/10.1152/ajpendo.1985.249.1.E43.
Johansson A, Lau J, Sandberg M, Borg LAH, Magnusson PU, Carlsson P-O. Endothelial cell signalling supports pancreatic beta cell function in the rat. Diabetologia. 2009;52:2385–94. https://doi.org/10.1007/s00125-009-1485-6.
Davalli AM, Scaglia L, Zangen DH, Hollister J, Bonner-Weir S, Weir GC. Vulnerability of islets in the immediate posttransplantation period. Dynamic changes in structure and function. Diabetes. 1996;45:1161–7. https://doi.org/10.2337/diabetes.45.9.1161.
de Groot M, Schuurs T, Fekken S, Leuvenink H, van Schilfgaarde R, Keizer J. Response of encapsulated rat pancreatic islets to hypoxia. Cell Transplant. 2003;12:867–75. https://doi.org/10.3727/000000003771000219.
Campbell PD, Weinberg A, Chee J, Mariana L, Ayala R, Hawthorne WJ, et al. Expression of pro- and antiapoptotic molecules of the Bcl-2 family in human islets postisolation. Cell Transplant. 2012;21:49–60. https://doi.org/10.3727/096368911X566262.
Cantley J, Walters SN, Jung M, Weinberg A, Cowley MJ, Whitworth PT, et al. A preexistent hypoxic gene signature predicts impaired islet graft function and glucose homeostasis. Cell Transplant. 2013;22:2147–59. https://doi.org/10.3727/096368912X658728.
Vajkoczy P, Menger MD, Simpson E, Messmer K. Angiogenesis and vascularization of murine pancreatic islet isografts. Transplantation. 1995;60:123–7. https://doi.org/10.1097/00007890-199507000-00002.
Mattsson G, Jansson L, Carlsson P-O. Decreased vascular density in mouse pancreatic islets after transplantation. Diabetes. 2002;51:1362–6. https://doi.org/10.2337/diabetes.51.5.1362.
Cho H-J, Lee N, Lee JY, Choi YJ, Ii M, Wecker A, et al. Role of host tissues for sustained humoral effects after endothelial progenitor cell transplantation into the ischemic heart. J Exp Med. 2007;204:3257–69. https://doi.org/10.1084/jem.20070166.
Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat Med. 2003;9:702–12. https://doi.org/10.1038/nm0603-702.
Peiris HS, Bonder CS, Coates PT, Keating DJ, Jessup CF. The b-cell/EC axis: how do islet cells talk to each other? Diabetes Care. 2014;63:3–11. https://doi.org/10.2337/db13-0617.
Kang S, Park HS, Jo A, Hong SH, Lee HN, Lee YY, et al. Endothelial progenitor cell cotransplantation enhances islet engraftment by rapid revascularization. Diabetes. 2012;61:866–76. https://doi.org/10.2337/db10-1492.
Quaranta P, Antonini S, Spiga S, Mazzanti B, Curcio M, Mulas G, et al. Co-transplantation of endothelial progenitor cells and pancreatic islets to induce long-lasting normoglycemia in streptozotocin-treated diabetic rats. PLoS One. 2014;9:1–13. https://doi.org/10.1371/journal.pone.0094783.
• Penko D, Rojas-Canales D, Mohanasundaram D, Peiris HS, Sun WY, Drogemuller CJ, et al. Endothelial progenitor cells enhance islet engraftment, influence beta cell function and modulate islet connexin 36 expression. Cell Transplant. 2015;24:37–48. https://doi.org/10.3727/096368913X673423 Co-transplantation of endothelial progenitor cells with pancreatic islets enhanced islet engraftment by improving the cure rate and the glucose control.
Golshayan D, Pascual M. Tolerance-inducing immunosuppressive strategies in clinical transplantation. Drugs. 2008;68:2113–30. https://doi.org/10.1016/S0140-6736(98)07493-5.
Xing Y, Hogquist KA. T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol 2012;1–16.
Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. https://doi.org/10.1038/nri2785.
Tang Q, Bluestone JA. Regulatory T-cell therapy in transplantation: moving to the clinic. Cold Spring Harb Perspect Med. 2013;3:1–15. https://doi.org/10.1101/cshperspect.a015552.
Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32. https://doi.org/10.1038/nri2343.
Cunningham EC, Sharland AF, Alex Bishop G. Liver transplant tolerance and its application to the clinic: can we exploit the high dose effect? Clin Dev Immunol. 2013;2013:1–9. https://doi.org/10.1155/2013/419692.
Cippà PE, Fehr T. Spontaneous tolerance in kidney transplantation - an instructive, but very rare paradigm. Transpl Int. 2011;24:534–5. https://doi.org/10.1111/j.1432-2277.2011.01260.x.
Orlando G, Hematti P, Stratta R, Burke G, Di Cocco P, Pisani F, et al. Clinical operational tolerance after renal transplantation. Ann Surg. 2010;252:915–28. https://doi.org/10.1038/nbt.3121.ChIP-nexus.
Li W, Kuhr CS, Zheng XX, Carper K, Thomson AW, Reyes JD, et al. New insights into mechanisms of spontaneous liver transplant tolerance: the role of Foxp3-expressing CD25+CD4+ regulatory T cells. Am J Transplant. 2008;8:1639–51. https://doi.org/10.1111/j.1600-6143.2008.02300.x.
Heidt S, Wood KJ. Biomarkers of operational tolerance in solid organ transplantation. Expert Opin Med Diagn. 2012;6:281–93. https://doi.org/10.1517/17530059.2012.680019.BIOMARKERS.
Niemann N, Sawitzki B. Treg therapy in transplantation: how and when will we do it ? Curr Transplant Reports. 2015;2:233–41. https://doi.org/10.1007/s40472-015-0066-5.
Riley JL, June CH, Blazar BR. Human T regulatory cells as therapeutic agents: take a billion or so of these and call me in the morning. Immunity. 2010;30:656–65. https://doi.org/10.1016/j.immuni.2009.04.006.Human.
McMurchy AN, Bushell A, Levings MK, Wood KJ. Moving to tolerance: clinical application of T regulatory cells. Semin Immunol. 2011;23:304–13. https://doi.org/10.1016/j.smim.2011.04.001.
Xiao F, Ma L, Zhao M, Huang G, Mirenda V, Dorling A, et al. Ex vivo expanded human regulatory T cells delay islet allograft rejection via inhibiting islet-derived monocyte chemoattractant protein-1 production in CD34+ stem cells-reconstituted NOD-scid IL2rgnull mice. PLoS One. 2014;9:1–12. https://doi.org/10.1371/journal.pone.0090387.
Wu DC, Hester J, Nadig SN, Zhang W, Trzonkowski P, Gray D, et al. Ex vivo expanded human regulatory T cells can prolong survival of a human islet allograft in a humanized mouse model. Transplantation. 2013;96:707–16. https://doi.org/10.1097/TP.0b013e31829fa271.
• Chandran S, Tang Q, Sarwal M, Polyclonal Regulatory T. Cell therapy for control of inflammation in kidney transplants. Am J Transplant. 2017;17:2945–54. https://doi.org/10.1111/ajt.14415 Infusion of ex vivo expanded regulatory T cells was well tolerated with no adverse effects in kidney transplant recipients.
• Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. 2016;64:632–43. https://doi.org/10.1002/hep.28459 Infusion of ex vivo expanded regulatory T cells was well tolerated with no adverse effects in liver transplant recipients. Seven patients have been maintaining normal graft function without the use of immunosuppressive drugs for more than 16 months.
Ackermann AM, Gannon M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J Mol Endocrinol. 2007;38:193–206. https://doi.org/10.1677/JME-06-0053.
Hughes A, Rojas-canales D, Drogemuller C. IGF2: an endocrine hormone to improve islet transplant survival. J Endocrinol. 2014;221:R41–8. https://doi.org/10.1530/JOE-13-0557.
Reik W, Sun F-L, Dean WL, Kelsey G, Allen ND. Transactivation of Igf2 in a mouse model of Beckwith-Wiedemann syndrome. Nature. 1997;389:809–15. https://doi.org/10.1038/39797.
Petrik J, Arany E, McDonald TJ, Hill DJ. Apoptosis in the pancreatic islet cells of the neonatal rat is associated with a reduced expression of insulin-like growth factor II that may act as a survival factor. Endocrinology. 1998;139:2994–3004. https://doi.org/10.1210/endo.139.6.6042.
Hill DJ, Strutt B, Arany E, Zaina S, Coukell S, Graham CF. Increased and persistent circulating insulin-like growth factor II in neonatal transgenic mice suppresses developmental apoptosis in the pancreatic islets. Endocrinology. 2000;141:1151–7. https://doi.org/10.1210/endo.141.3.7354.
Hogg J, Han VK, Clemmons DR, Hill DJ. Interactions of nutrients, insulin-like growth factors (IGFs) and IGF-binding proteins in the regulation of DNA synthesis by isolated fetal rat islets of Langerhans. J Endocrinol. 1993;138:401–12. https://doi.org/10.1677/joe.0.1380401.
Robitaille R, Dusseault J, Henley N, Rosenberg L, Hallé J-P. Insulin-like growth factor II allows prolonged blood glucose normalization with a reduced islet cell mass transplantation. Endocrinology. 2003;144:3037–45. https://doi.org/10.1210/en.2002-0185.
Petrik J, Reusens B, Arany E, Remacle C, Coelho C, Hoet JJ, et al. A low protein diet alters the balance of islet cell replication and apoptosis in the fetal and neonatal rat and is associated with a reduced pancreatic expression of insulin-like growth factor-II. Endocrinology. 1999;140:4861–73. https://doi.org/10.1210/endo.140.10.7042.
Hughes A, Mohanasundaram D, Kireta S, Jessup CF, Drogemuller CJ, Coates PTH. Insulin-like growth factor-II (IGF-II) prevents proinflammatory cytokine-induced apoptosis and significantly improves islet survival after transplantation. Transp J. 2013;95:671–8. https://doi.org/10.1097/TP.0b013e31827fa453.
Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes and Cancer. 2011;2:1097–105. https://doi.org/10.1177/1947601911423031.
Lammert E, Gu G, Mclaughlin M, Brown D, Brekken R, Murtaugh LC, et al. Role of VEGF-A in vascularization of pancreatic islets. Curr Biol. 2003;13:1070–4. https://doi.org/10.1016/S0960-9822(03)00378-6.
Witkowski P, Sondermeijer H, Hardy MA, Woodland DC, Lee K, Bhagat G, et al. Islet grafting and imaging in a bioengineered intramuscular space. Transplantation. 2009;88:1065–74. https://doi.org/10.1097/TP.0b013e3181ba2e87.
• Marchioli G, Di Luca A, De Koning E, Engelse M, Van Blitterswijk CA, Karperien M, et al. Hybrid polycaprolactone/alginate scaffolds functionalized with VEGF to promote de novo vessel formation for the transplantation of islets of Langerhans. Adv Healthc Mater. 2016;5:1606–16. https://doi.org/10.1002/adhm.201600058 Polycaprolactone scaffold functionalized with VEGF promoted vascularization and improved function of encapsulated islets embedded within the polycaprolactone scaffold.
Boyman O, Sprent J. The role of interleukin - 2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12:180–90. https://doi.org/10.1038/nri3156.
Sakaguchi S, Takahashi T, Nishizuka Y. Study on cellular events in post-thymectomy autoimmune oophoritis in mice. J Exp Med. 1982;156:1577–86.
Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for IL-2 receptor in regulatory T cell function Takatoshi. Nat Immunol. 2016;17:1322–33. https://doi.org/10.1038/ni.3540.
Létourneau S, Krieg C, Pantaleo G, Boyman O. IL-2- and CD25-dependent immunoregulatory mechanisms in the homeostasis of T-cell subsets. J Allergy Clin Immunol. 2009;123:758–62. https://doi.org/10.1016/j.jaci.2009.02.011.
Sadlon TJ, Wilkinson BG, Pederson S, Brown CY, Bresatz S, Gargett T, et al. Genome-wide identification of human FOXP3 target genes in natural regulatory T cells. J Immunol. 2010;185:1071–81. https://doi.org/10.4049/jimmunol.1000082.
Beyer M, Thabet Y, Müller R, Sadlon T, Classen S, Lahl K, et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol. 2011;12:898–907. https://doi.org/10.1038/ni.2084.
Whitehouse G, Gray E, Mastoridis S, Merritt E, Kodela E, Yang JHM. IL-2 therapy restores regulatory T-cell dysfunction induced by calcineurin inhibitors. Proc Natl Acad Sci USA. 2017;114:7083–8. https://doi.org/10.1073/pnas.1620835114.
Shevach EM. Application of IL-2 therapy to target T regulatory cell function. Trends Immunol. 2013;33:626–32. https://doi.org/10.1016/j.it.2012.07.007.
Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+ CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–80. https://doi.org/10.1182/blood-2006-02-004747.
Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365:2055–66. https://doi.org/10.1056/NEJMoa1108188.
Matsuoka K, Koreth J, Kim HT, Bascug OG, Kawano Y, Murase K, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med. 2013;5:179ra43. https://doi.org/10.1126/scitranslmed.3005265.
Koreth J, Kim HT, Jones KT, Lange PB, Reynolds CG, Chammas MJ, et al. Efficacy , durability , and response predictors of low-dose interleukin-2 therapy for chronic graft-versus-host disease. Blood. 2016;128:130–8. https://doi.org/10.1182/blood-2016-02-702852.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Immunology, Transplantation, and Regenerative Medicine
Rights and permissions
About this article

Cite this article
Kim, J., Kang, K., Drogemuller, C.J. et al. Bioprinting an Artificial Pancreas for Type 1 Diabetes. Curr Diab Rep 19, 53 (2019). https://doi.org/10.1007/s11892-019-1166-x
Published:
DOI: https://doi.org/10.1007/s11892-019-1166-x