
Abstract
Genome-wide association studies for coronary artery disease utilizing the case control association study approach has identified 50 genetic risk variants associated with coronary artery disease or myocardial infarction. All of these genetic variants are of genome wide significance and replicated in an independent population. It is of note that 35 of these 50 genetic risk variants act through mechanisms as yet unknown. These findings have great implications for the pathogenesis of atherosclerosis, as well as new targets for the development of novel therapies for the prevention and treatment of CAD. The genetic variant PCSK9 has already led to the development of a monoclonal anti-body which is undergoing assessment in phases I, II, and III clinical trials. This therapy shows very promising results and since it increases removal of LDL-C, it is complementary to current statin therapy. Assessing the beneficial or deleterious effects of a lifelong exposure to a genetic risk variant (Mendelian randomization) will be an important adjunct to clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Over the last four decades, attempts to define the genetics of coronary artery disease have been restricted to that of rare single gene disorders, primarily involving cholesterol. The most striking observation of that era was by Brown and Goldstein describing a mutation in the low-density lipoprotein receptor responsible for familial hypercholesterolemia [1]. Only in the last decade has it been possible to pursue genetic predisposition underlying complex multi-gene disorders, such as coronary artery disease (CAD). This was heralded by the development of high-density microarrays containing hundreds of thousands of single nucleotide polymorphisms (SNPs) as DNA markers and the requisite platforms to perform unbiased genome-wide association study (GWAS) [2]. This review focuses on this approach and its contribution to the genetic architecture that underlies CAD.
Genome-Wide Association Studies (GWAS) for Coronary Artery Disease
Genetic linkage analysis provided the resolution required to map and identify genes responsible for rare disorders such as familial hypercholesterolemia or familial hypertrophic cardiomyopathy. It required genotyping with only a few hundred DNA markers in pedigrees of two or three generations [3]. One can map the chromosomal location of the responsible gene by detecting the markers that segregate with the disease versus those that segregate with unaffected individuals. Markers that are co-inherited with the disease would indicate they are in close physical proximity to that of the responsible gene. Knowing the approximate chromosomal location, one can, through cloning and sequencing, identify the precise mutation. It was recognized in the 1990s that while this approach would be applicable for single gene disorders, it would not have the resolution for complex diseases in which the phenotype results from a combination of multiple genetic and environmental factors. Knowing there are multiple genetic factors contributing, it was postulated that the effect of any one genetic factor would have minimal to moderate effect on the phenotype. This would require an approach utilizing unrelated individuals best suited to the case control association study. In this approach, one compares the frequency of a DNA marker in cases versus the frequency in controls. A marker occurring statistically more frequent in cases, would be interpreted to be in a region of DNA associated with increased risk for the disease. The initial approach taken in the 1990s was to select candidate genes on the basis of their function and compare their frequency in cases and controls. While several such studies were performed for many diseases including CAD, there was the concern of the built in bias from prior selection of the candidate [4]. The most desired approach would be to have hundreds of thousands of markers evenly distributed throughout the genome and chose those markers occurring more frequently in cases without any prior bias. To have adequate resolution, such an approach is now referred to as a genome-wide association study (GWAS), would require a DNA marker at intervals throughout the genome of at least every 6000 base pairs of the 3.2 billion base pairs present in the genome [2]. This would require a minimum of 500,000 markers and sample sizes for both cases and controls of several thousand individuals.
The development of the high-density microarray containing initially 500,000 SNPs and subsequently 1 million single nucleotide polymorphisms (SNPs) facilitated the first complete unbiased GWAS [2]. Given the number of hypotheses being tested, a statistical correction is necessary to account for false positives. The conventional accepted statistical correction for genome significance is the Bonferroni method using <0.05/1,000,000 = 5 × 10-8 [5]. To meet this statistical stringency would require large sample sizes of several thousand. The resolution afforded by a GWAS is to detect SNPs occurring at a frequency of ≥5 % in the population. Even with massive sample sizes, GWAS does not have the resolution to detect SNPs having a frequency of <5 %. As these findings are based on a case-control association study by comparing the frequency of a SNP in cases with that of controls, it is only an association and must be replicated in an independent and appropriate population. Thus far 50 genetic variants associated with risk of CAD have met this degree of stringency.
9p21 Risk Variant and Coronary Artery Disease
The initial observation that emerged from GWAS for CAD was the discovery of an association between a DNA sequence variation on chromosome 9p21 and coronary artery disease (CAD) [6, 7]. Since its initial description in 2007, associations between 9p21 risk variant and other vascular phenotypes have been described, including intracranial aneurysms, intra-abdominal aortic aneurysms and stroke [8]. The 9p21 risk variant is the most studied and typifies most of the features of the newly discovered genetic risk variants for CAD.
The 9p21 risk variant for CAD is very common with 25 % of the Caucasian population being homozygous for the risk variant and 50 % of the population heterozygous [6]. Thus 75 % of the Caucasian population carries at least one copy of the risk variant. While the risk imparted is modest, the high frequency of 9p21 led to its confirmation in multiple GWAS. The association has subsequently been described in multiple ethnic groups including the Japanese [9], Korean [9, 10], and South Asian [11]. However, the association has not been replicated in African American populations [12]. Somewhat surprisingly, and of great therapeutic interest, was the observation that the risk of 9p21 for CAD is independent of known risk factors for CAD (e.g., cholesterol).
The 9p21 risk variant resides in a region of the genome devoid of annotated protein coding genes. The risk variant resides in a long non-coding RNA (lncRNA) of 126,000 bps, antisense non-coding RNA at the INK4 locus (ANRIL). This sequence lies adjacent to the genes that encode the cyclin-dependent kinase inhibitors CDKN2A and CDKN2B [13]. In mice targeted deletion of the analogous DNA region results in reduced expression of CDKN2A [14], however, these mice did not develop an atherosclerotic phenotype. The CDKN2A and CDKN2B are inhibitors of the cell cycle and are known to inhibit cellular proliferation. Since smooth muscle proliferation is part of atherosclerosis, inhibition of their expression may enhance proliferation of atherosclerosis. Of note, the murine sequence shares only 50 % homology with that of humans [14]. In vitro and in vivo studies show inconsistent results with respect to whether the 9p21 risk variant is associated with mRNA expression of CDKN2A and CDKN2B [13, 15, 16]. Genetic transmission of genes is random and each gene is thus inherited independently. However, genes that are in close physical proximity may be co-inherited and are referred to as being in linkage disequilibrium.
Following sequencing of the region, other SNPs that are in linkage disequilibrium with the original CAD-risk SNPs have been identified. Two of these SNPs exist in a binding site for the transcription factor STAT1, a transcription factor induced by interferon-gamma [17•]. The haplotype that contains SNPs conferring CAD risk disrupt STAT1 binding; knockdown of STAT1 by short interfering RNAs increased CDKN2BAS expression in cells homozygous for the non-risk allele but not in those homozygous for the risk allele (i.e., with disrupted STAT1 biding sites) [17•]. This led to the hypothesis that 9p21 may mediate its risk for CAD through interferon gamma [17•]; however, it was subsequently shown that the effect of interferon gamma is independent of the 9p21 risk variant [18•]. The mechanism by which 9p21 mediates its risk is yet to be elucidated.
While the mechanism whereby 9p21 mediates its risk remains unknown, its site of action is more clearly elucidated. Studies in patients having undergone coronary angiography with or without myocardial infarction (MI) have shown the 9p21 risk variant to be associated with coronary atherosclerosis but not with MI [19]. This has been consistently observed in multiple studies [20–22]. Several studies have also documented 9p21 risk variant to be associated with progression of coronary atherosclerosis [19, 22, 23] based on the number of vessels involved, however, this has not been a consistent finding [9, 21]. All of these studies are cross-sectional analysis, while it may require a longitudinal study to definitively solve this controversy. It is reasonable to conclude that 9p21 risk variant acts at the vessel wall in association with coronary atherosclerosis and is not associated with plaque rupture or thrombosis.
Common Features of Genetic Risk Variants for CAD
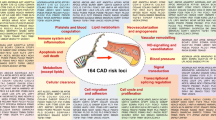
Following the description of the association of the 9p21 risk variant with coronary artery disease, several other variants were described [3]. Given the modest risk effect of these genetic variants, it was recognized that sample sizes would have to be larger than initially expected. As a consequence, members of several international consortia contributed patients and resources to a large meta-analysis. The largest of these consortia is Coronary Artery Disease genome-wide Replication and Meta-analysis (CARDIoGRAM) [24]. This study initially included 87,000 subjects, of whom 22,000 were CAD cases and 65,000 controls. CARDIoGRAM identified 13 new risk variants for CAD while confirming ten others that had been previously described [25]. CARDIoGRAM expanded its population to over 200,000 and recently identified another 15 risk variants with confirmation of a total of 46 [26••]. Four other genetic risk variants for CAD from independent investigators have also been identified [27, 28]. A total of 50 genetic risk variants for CAD have been discovered and confirmed in independent populations (Table 1).
Several features have now emerged that are common to all of these genetic risk variants for CAD. They are very common with over 50 % of them occurring in 50 % of the population and over 30 % occurring in 75 % or more of the population (Table 1). Secondly, the extent of increased risk for any one genetic variant is minimal, with an average increased relative risk of 17 %. Most of the genetic variants occur in non-protein coding regions, indicating their effect is regulatory, acting on downstream or upstream protein coding regions. Their effect on DNA regions on other chromosomes must also be considered. Perhaps the most important finding is that 35 of the 50 genetic risk variants act independent of known risk factors by mechanisms that are, as of yet, unknown.
Myocardial Infarction Versus Coronary Artery Disease
In all GWAS studies, the phenotype is either confirmed myocardial infarction or the presence of ≥50 % obstruction in one or more of the coronary vessels. The two phenotypes are treated as similar since a myocardial infarction is essentially always due to plaque rupture and thrombosis superimposed on underlying obstructive coronary artery atherosclerosis. Nevertheless, it is expected that certain genetic risk variants will mediate their risk solely through myocardial infarction or atherosclerosis. It is reasonable to expect there are separate genetic variants predisposing to thrombosis, such as the ABO locus that does not contribute to atherosclerosis per se. Similarly, genetic variants predisposing to plaque rupture may be distinct from those associated with the pathogenesis of atherosclerosis or the formation of thrombosis. It is worth noting that only one genetic risk variant was observed to be associated with MI per se, namely, the blood group locus occupied by either A, B, or O (Table 1). Epidemiological studies since the 1960s have indicated increased risk for MI is associated with blood group A and B, the GWAS studies confirmed that A and B have a relative increased risk of about 20 % for MI, but blood group O was not associated with any risk for CAD or MI [20]. The genes A or B encode for a transferase that transfers a carbohydrate moiety onto von Willebrand’s factor. The longer half-life predisposes to thrombosis and would account for the increased risk for MI associated with A and B blood groups. However, blood group O while it encodes for the same transferase has been mutated such that the enzyme now lacks activity resulting in no change in the half-life of von Willebrand’s factor and thus imparts no increased risk for MI. In the recent nurses population study [29], it was confirmed that blood group A or B associated with increased risk of about 10 %, while if you have two copies of A or B or AB, the risk is increased by about 20 %. It has also been shown that in individuals with blood group A or B, the plasma half-life of the vWF is prolonged by about 25 % [30]. While there are no current recommendations on how to manage individuals with blood group A or B, future concern will be related as to whether they should receive anti-platelet therapy following coronary by-pass surgery or the insertion of permanent artificial devices such as stents or prosthetic valves.
Utility of Genetic Risk Variants in the Prevention of CAD
While identification of the novel variants is of scientific interest and identification of the mode by which they act will likely yield valuable insights with regard to pathogenesis of disease, the value of their application in prevention will depend on demonstrating genotyping improves risk prediction above and beyond that achieved by standard risk-prediction models.
Ripatti et al. investigated genetic risk scoring based on the first 13 SNPs described in GWAS [31]. They found this genetic score identified 20 % of a European population that were at a 70 % increased risk of a first coronary event. However, genetic risk scoring did not improve the C index over traditional risk factors and family history nor did it have an effect on net reclassification improvement.
Thanassoulis et al. used genetic risk scoring (13 SNPs) in the prediction of coronary artery calcium (CAC) and incident CHD [32]. Consistent with Ripatti’s observation, genetic risk scoring did not improve discrimination for incident CAD. However, significant improvements in risk discrimination and reclassification were observed in the prediction of high CAC.
Benefits Emerging from Discovery of Genetic Risk for CAD
While the search for addition genetic risk variants for CAD continues, a major finding has already immerged, namely, that several factors contributing to the pathogenesis of atherosclerosis are yet to be discovered. Over two-thirds of the genetic risk variants for CAD mediate their effect through mechanisms other than cholesterol or blood pressure (Table 1). The molecular pathways are yet to be elucidated. These pathways will provide new targets for the development of novel therapy. It is now evident that comprehensive prevention and treatment of coronary atherosclerosis and its sequelae must include therapy to counteract these genetic risk factors.
In Table 1 it is indicated that seven of the genetic risk variants are associated with plasma LDL-cholesterol. One of these variants is due to mutations in PCSK9 located on chromosome 1 (1p32.3). PCSK9 increases the degradation of the LDL receptor [33]. Since the major mechanism responsible for removal of LDL-C from the plasma is the LDL receptor, it is associated with increased plasma LDL-C. Loss of function mutations in PCSK9 was shown to be associated with lower plasma levels of LDL-C and decreased mortality and morbidity from CAD [34]. An anti-body inhibiting the action of PCSK9 has undergone evaluation in phase I and II clinical trials, with very promising results, which are now being further evaluated in phase III clinical trials [35–37]. This therapy has been shown to be complementary to that of statins, as expected since statin therapy inhibits the synthesis of cholesterol while inhibiting PCSK9 results in more rapid removal of LDL-C from the plasma. One can expect other such novel therapies to lower plasma cholesterol will emerge. It is reasonable to expect that significant insight will be obtained from the homeostasis of cholesterol through pursuing the actions of these new genetic variants related to the regulation of plasma lipids and the risk of CAD
Conclusion
GWAS has led to the description of multiple novel CAD loci. This has improved our understanding of the genetic architecture that underlies CAD and its related phenotypes. These variants account for only about 20 % of expected CAD heritability. Future directions will include 1) description of novel variants through larger meta-analyses, the identification of rare variants that impart profound biologic effect by techniques including next-generation sequencing and whole-exome capture; 2) and description of the mechanisms that underlie the association of these SNPs with CAD; 3) re-assessment of the role of genotyping in risk prediction following description of further variants.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Brown MS, Goldstein JL. Familial hypercholesterolemia: a genetic defect in the low-density lipoprotein receptor. N Engl J Med. 1976;294(25):1386–90.
Roberts R. New gains in understanding coronary artery disease, interview with Dr. Robert Roberts. Affymetrix Microarray Bull. 2007;3(2):1–4. Spring.
Dandona S et al. Genomics in coronary artery disease: past, present and future. Can J Cardiol. 2010;26(Suppl A):56A–9A.
Pare G. Genetic analysis of 103 candidate genes for coronary artery disease and associated phenotypes in a founder population reveals a new association between endothelin-1 and high-density lipoprotein cholesterol. Am J Hum Genet. 2007;80(4):673–82.
Samani NJ et al. Genome wide association analysis of coronary artery disease. N Engl J Med. 2007;357(5):443–53.
McPherson R et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316(5830):1488–91.
Helgadottir A et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316(5830):1491–3.
Helgadottir A et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40(2):217–24.
Hinohara K et al. Replication of the association between a chromosome 9p21 polymorphism and coronary artery disease in Japanese and Korean populations. J Hum Genet. 2008;53(4):357–9.
Shen GQ et al. Four SNPs on chromosome 9p21 in a South Korean population implicate a genetic locus that confers high cross-race risk for development of coronary artery disease. Arterioscler Thromb Vasc Biol. 2008;28(2):360–5.
Kumar J et al. Association of polymorphisms in 9p21 region with CAD in North Indian population: replication of SNPs identified through GWAS. Clin Genet. 2011;79(6):588–93.
Roberts R, Stewart AF. Genes and coronary artery disease: where are we? J Am Coll Cardiol. 2012;60(18):1715–21.
Kim JB et al. Effect of 9p21.3 coronary artery disease locus neighboring genes on atherosclerosis in mice. Circulation. 2012;126(15):1896–906.
Visel A et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464(7287):409–12.
Jarinova O et al. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. 2009;29(10):1671–7.
Burd CE et al. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6(12):e1001233.
Harismendy O et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470(7333):264–8. Proposed that interferon-gamma mediates the risk of 9p21 for CAD.
Almontashiri NA et al. Interferon-y activates expression of p15 and p16 regardless of 9p21.3 coronary artery disease risk genotype. J Am Coll Cardiol. 2013;61(2):143–7. These studies indicate the effects of interferon-gamma on expression of P15 and P16 are independent of the 9p21 risk allele.This does not support the hypothesis of 9p21 by Harismendy.
Dandona S et al. Gene dosage of the common variant 9p21 predicts severity of coronary artery disease. J Am Coll Cardiol. 2010;56(6):479–86.
Reilly MP et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377(9763):383–92.
Horne BD et al. Association of variation in the chromosome 9p21 locus with myocardial infarction versus chronic coronary artery disease. Circ Cardiovasc Genet. 2008;1(2):85–92.
Ardissino D et al. Influence of 9p21.3 genetic variants on clinical and angiographic outcomes in early-onset myocardial infarction. J Am Coll Cardiol. 2011;58(4):426–34.
Chan K et al. Association between the chromosome 9p21 locus and angiographic coronary artery disease burden: a collaborative meta-analysis. J Am Coll Cardiol. 2013;61(9):957–70.
Preuss M et al. Design of the Coronary ARtery DIsease Genome-Wide Replication and Meta-Analysis (CARDIoGRAM) Study: a Genome-wide association meta-analysis involving more than 22 000 cases and 60 000 controls. Circ Cardiovasc Genet. 2010;3(5):475–83.
Schunkert H et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43(4):333.8.
Deloukas P, CARDIoGRAMplusC4D Consortium, et al. Large-scale association analysis identified new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33. This study involving over 240,000 individuals discovers 15 new genetic risk variants for CAD, and confirms 31 previous genetic risk variants for a total of 46. This study indicates all of the genetic variants are associated with CAD, having a P value of genome wide significance and the association has been replicated in an independent population.
Lu X et al. Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet. 2012;44(8):890–4.
IBC 50 K CAD Consortium. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011;7(9):e1002260.
He M et al. ABO blood group and risk of coronary heart disease in two prospective cohort studies. Arterioscler Thromb Vasc Biol. 2012;32(9):2314–20.
Gill JC et al. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood. 1987;69(6):1691–5.
Ripatti S et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet. 2010;376(9750):1393–400.
Thanassoulis G et al. A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: the Framingham Heart Study. Circ Cardiovasc Genet. 2012;5(1):113–21.
Roberts R. PCSK9 inhibition - a new thrust in the prevention of heart disease: genetics does it again. Can J Cardiol. 2013;29(8):899–901.
Cohen JC et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–72.
Wierzbicki AS et al. Inhibition of pro-protein convertase subtilisin kexin 9 [corrected] (PCSK-9) as a treatment for hyperlipidaemia. Expert Opin Investig Drugs. 2012;21(5):667–76.
Stein EA et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J Med. 2012;366(12):1108–18.
Ni YG et al. A PCSK9-binding antibody that structurally mimics the EGF(A) domain of LDL-receptor reduces LDL cholesterol in vivo. J Lipid Res. 2011;52(1):78–86.
Acknowledgments
In the preparation of this document, the authors would like to acknowledge the efforts of Peggy Offley and Heather Stevenson.
Robert Roberts has received grant support from CIHR #MOP82810 Canada, CFI # 11966 Canada.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Sonny Dandona declares that he has no conflict of interest.
Robert Roberts is a consultant to Cumberland Pharmaceuticals.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Diabetes and Cardiovascular Disease
Rights and permissions
About this article
Cite this article
Dandona, S., Roberts, R. The Role of Genetic Risk Factors in Coronary Artery Disease. Curr Cardiol Rep 16, 479 (2014). https://doi.org/10.1007/s11886-014-0479-2
Published:
DOI: https://doi.org/10.1007/s11886-014-0479-2