
Abstract
Lipodystrophies are rare acquired and genetic disorders characterized by the selective loss of adipose tissue. One key metabolic feature of patients with congenital inherited lipodystrophy is hypertriglyceridemia. The precise mechanisms by which the lack of adipose tissue causes dyslipidemia remain largely unknown. In recent years, new insights have arisen from data obtained in vitro in adipocytes, yeast, drosophila, and very recently in several genetically modified mouse models of generalized lipodystrophy. A common metabolic pathway involving accelerated lipolysis and defective energy storage seems to contribute to the dyslipidemia associated with congenital generalized lipodystrophy syndromes, although the pathophysiological changes may vary with the nature of the mutation involved. Therapeutic management of dyslipidemia in patients with lipodystrophy is primarily based on specific approaches using recombinant leptin therapy. Preclinical studies suggest a potential efficacy of thiazolidinediones that remains to be assessed in dedicated clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipodystrophies regroup a heterogeneous group of syndromes in which the primary defect is a lack of adipose tissue that may be congenital or acquired in origin [1••, 2, 3] (summarized in Table 1). Lipodystrophy should be considered in patients manifesting the combination of insulin resistance (with or without overt diabetes), dyslipidemia (mainly hypertriglyceridemia), and liver steatosis [4]. Lipodystrophies are classified according to the origin of the disease (genetic or acquired) and the anatomical distribution of adipose tissue (generalized or partial). The severity of the metabolic phenotype is about proportional to the degree of adipose tissue loss, and therefore tends to be worse in generalized lipodystrophy. In this review, we will focus on the clinical characteristics, molecular mechanisms, and treatment of dyslipidemias related to congenital lipodystrophies, with special mention of Berardinelli–Seip congenital lipodystrophy (BSCL).
Classification of Congenital Lipodystrophies and Clinical Characterization
Congenital Generalized Lipodystrophies (BSCL)
BSCL is characterized by an almost complete lack of adipose tissue from birth or early infancy [1••, 2, 3, 5]. Metabolic complications generally appear progressively during childhood. Early insulin resistance (reflected by the presence of acanthosis nigricans) progresses to overt diabetes during the teenage years or later. Plasma leptin and adiponectin concentrations are reduced in correlation with the amount of residual fat [6], although in BSCL type 2, a surprisingly detectable adiponectin level has been reported [7].
BSCL is a rare heterogeneous recessively inherited disorder. Positional cloning strategies have led to the identification of two major loci: BSCL1 on chromosome band 9q34 [8] encoding acylglycerol-3-phosphate-O-acyltransferase 2 (AGPAT2) [9] and BSCL2 on chromosome band 11q13 encoding seipin [10], which are involved in most cases [11]. In a few cases, other BSCL-causative genes have been identified: CAV1 (BSCL3) encoding caveolin 1 has been reported in single patient [12] and PTRF (BSCL4) encoding polymerase I and transcript release factor also known as cavin 1 [13, 14] has been reported in about 20 patients. Recently, a mutation in the FOS (encoding the transcription factor c-Fos) promoter has been identified in a single patient, in whom the phenotype and the mode of inheritance are not well characterized [15]. It is likely that additional loci exist, as some patients with BSCL do not harbor mutations in any of the known genes.
Partial Lipodystrophies
Partial lipodystrophies are characterized by loss of fat affecting the limbs, with variable truncal involvement and normal or excess fat on the face and neck [1••, 2–4]. The onset of lipodystrophy usually occurs during childhood or puberty or later. Metabolic features range from asymptomatic impaired glucose tolerance with mild dyslipidemia to severe insulin resistance, diabetes, and dyslipidemia, depending on the extent of fat tissue loss. Nonalcoholic fatty liver disease (NAFLD) and cardiovascular disease are common complications.
The familial partial lipodystrophy (FPL) syndromes are usually transmitted according to an autosomal dominant mode of inheritance [16]. A positional cloning strategy led to the identification of the first locus for the Dunnigan variety of FPL, i.e., LMNA, on chromosome locus 1q21-22 [17–19]. LMNA encodes type A lamins, which are integral components of nuclear lamina. The candidate gene approach has led to the identification of four other FPL-causative genes: PPARG, encoding peroxisome-proliferator-activated receptor (PPAR) γ [20, 21], a key transcription factor involved in adipocyte differentiation; AKT2, encoding v-AKT murine thymoma oncogene homolog 2 [22], involved in insulin signaling; and CIDEC, encoding cell-death-inducing DNA fragmentation factor α like C [23], and PLIN, encoding perilipin 1 [24], both involved in lipid droplet (LD) maintenance (Table 1).
Clinical Characteristics of Dyslipidemia in Inherited Lipodystrophies
Hypertriglyceridemia is a common feature of BSCL, although it is variable among individuals within the same family and also within the same individual over time depending on age, food, and environmental factors such as physical activity [5, 25]. Such hypertriglyceridemia results from the accumulation of both chylomicrons and very low density lipoproteins (VLDL), and may lead to eruptive xanthomas and acute pancreatitis. Plasma levels of phospholipids and total cholesterol are usually within the normal range or are slightly elevated, whereas high-density lipoprotein (HDL) levels are decreased. However, increased plasma total cholesterol levels have sometimes been reported [12, 15]. Free fatty acids (FFA) levels are variable, normal, or slightly elevated. Hepatomegaly is a constant feature of BSCL [1••, 2, 3, 5, 25]. Accumulation of liver triglycerides (TGs) leads to hepatic steatosis and to increased VLDL synthesis, which contributes, to hypertriglyceridemia [26].
In vivo mechanistic studies are few in number and are confined mostly to individual case studies (with unknown genetic origin). For instance, in a 25-year-old patient with BSCL, the ability of insulin to inhibit lipolysis was markedly blunted [27]. In six patients with BSCL, the postprandial lipid metabolism evaluated using labeled TG-rich lipoprotein (TRL) emulsion revealed that both lipoprotein lipase (LPL)-mediated lipolysis and removal of TRL remnant were impaired and delayed [28]. The data on LPL activity are quite variable since in some reports LPL activity was reported to be normal [29, 30], whereas it was markedly reduced in other cases [31]. In striking contrast, plasma TG lipolysis appeared increased and uncontrolled in patients carrying the PPARγ P467L mutation [32]. Although P467L mutation causes partial lipodystrophy, these findings brought important insights into the pathological mechanisms of lipodystrophies. Postprandial FFA concentrations were increased in these patients and paralleled the peak of TG concentrations in the postprandial state. However, this rise in FFA concentrations was not linked to increased lipolysis, but rather to the inability of the tissue to internalize and store the 13C-palmitic acid coming from labeled TRL. Similar studies with BSCL patients of various genotypes will be necessary to establish if this is also the case in these patients. Nonetheless, a strong body of evidence suggests that adipose tissue failure strongly impairs the postprandial TG metabolism, owing to defective TG uptake. Indeed, lipodystrophy is associated with the inability to store excess of energy, since BSCL patients fed a high-fat diet over a period of time display increased plasma TG and FFA levels [29, 30]. When seven patients with different types of lipodystrophies were fed 30 % excess energy as fat, there was an increase in energy expenditure as a result of an increase in fat oxidation [33].
Mouse Models as Tools to Decipher the Molecular Mechanisms Involved in Dyslipidemia Related to Lipodystrophies
As human studies are difficult to conduct, the development of mouse models has provided key tools to improve our understanding of the molecular mechanisms involved in the origin of lipodystrophy and also important models to study the metabolic BSCL phenotypes (reviewed in [34]).
Inability to expand adipose tissue results in metabolic complications. The first proof of this concept in a mouse model arose from transgenic mice that express diphtheria toxin A under the adipose-tissue-specific AP2 promoter, resulting in a massive loss of adipose tissue. The inability to store TG in the adipose tissue resulted in a twofold increase in plasma TG levels and liver weight. No significant changes in cholesterol levels was reported. Even if this was not per se a model of lipodystrophy, this was the first publication that stated that in mice, limitation of the expansion of adipose tissue results in hypertriglyceridemia and liver steatosis [35].
Transcription Factors of Adipogenesis and Lipogenesis
Five years later, the two first mouse models of general lipodystrophy were generated. The overexpression of the nuclear mature form of the transcription factor SREBP1c under the AP2 promoter results in an alteration of the expression of key adipogenic factors, such as PPARγ and CCAAT/enhancer-binding protein α (C/EBPα), and results in a severe lipodystrophic phenotype, similar to human BSCL [36]. Insulin resistance and diabetes in these mice were associated with liver steatosis and hypertriglyceridemia. Notably, the plasma total cholesterol level was also increased in this model.
On the other hand, A-ZIP/F mice specifically express in adipose tissue, under the AP2 promoter, a dominant negative protein that binds to C/EBPα, a key transcription factor involved in adipogenesis [37]. A-ZIP/F mice are severely lipodystrophic and display a threefold increase in plasma TG levels and liver steatosis, suggesting that the lack of C/EBPα activity in the adipocyte was largely the cause of the lipodystrophic phenotype. Indeed, the lack of C/EBPα expression in adipose tissue was sufficient to induce a massive loss of white adipose tissue (WAT), a fatty liver, and a severe hypertriglyceridemia [38]. WAT transplantation in A-ZIP/F mice improves glucose and insulin levels but has a modest effect on TG levels. Leptin levels were improved by WAT transplantation in a dose-dependent manner, and the highest plasma leptin concentration coincided with the strongest effect on TG levels [39]. Consistently, leptin treatment of AP2-nSREBP1c mice reversed insulin resistance [40], and hepatic overexpression of leptin in an A-ZIP/F background corrects hypertriglyceridemia [41]. These data suggest that leptin levels play a key role in TG homeostasis in lipodystrophic mouse models.
PPARγ is the master regulator of adipogenesis, and embryonic stem cells lacking PPARγ cannot undergo adipogenesis [42]. Global PPARγ deletion is lethal [42, 43]. Adipose-tissue-specific deletion of PPARγ under the AP2 promoter results in mild and progressive lipodystrophy without a change in plasma TG levels [44]. In contrast, using the adiponectin promoter, Wang et al. [45•] demonstrated that the ablation of PPARγ in adipocytes leads to a severe and almost complete lipodystrophic phenotype. The mice were strongly hypoleptinemic, hypoadiponectinemic, and insulin-resistant, with a massive liver steatosis and a fivefold increase in plasma TG levels. In conclusion, genetic impairment of adipogenesis compromises adipose tissue development and homeostasis, resulting in severe lipodystrophy associated with massive liver steatosis and hypertriglyceridemia. Together, these findings reinforce the central role of adipose tissue in TG homeostasis.
Caveolae
Caveolae are specialized invaginations of the plasma membrane that are found in numerous cell types and that are particularly abundant in adipocytes. In 2001, Razani et al. [46] highlighted that caveolin-1-deficient mice were resistant to a high-fat diet and displayed fat loss and adipocyte abnormalities with ageing. Caveolin 1 is one of the key component of caveolae [47]. Following up this publication, several groups intended to understand why caveolin 1 deficiency leads to lipodystrophy with ageing. Despite unchanged messenger RNA levels, the protein levels of the insulin receptor are dramatically reduced in Cav1 -/- mice fat pads, altering therefore insulin signaling and sensitivity in this tissue [48]. In addition, caveolin 1 localizes to LDs in adipocytes [49] and its translocation to the LD can be rapidly induced by cholesterol incubation in 3 T3-L1 adipocyte cell lines [50]. Caveolin 1 deficiency strongly affects phospholipid composition and the morphology of LDs isolated from primary adipocytes [51]. Finally, caveolin 1 deficiency constitutively activates the autophagic process [52] and increases the susceptibility to cell death [53]. Cav1 -/- mice are hypertriglyceridemic in the fasting and postprandial states and display strong delay in postprandial TG clearance [46]. LPL activity is not affected, and the origin of this hypertriglyceridemia is mainly attributed to the inability of caveolin-1-deficient adipocytes to store lipids properly in the LDs. Since caveolin 1 is a cholesterol-binding protein, the consequences of its deletion on plasma cholesterol metabolism have been studied. Caveolin 1 deficiency has no impact on cholesterol levels on a wild-type background, but does lead to an increase in low density lipoprotein (LDL) cholesterol levels on an ApoE -/- background [54]. Intriguingly, this rise in LDL cholesterol levels was associated with a decrease in the area of atherosclerotic lesions, as CD36 expression is strongly decreased in caveolin-1-deficient macrophages. It remains unclear, however, whether lipodystrophy per se is involved in the hypercholesterolemia reported in these mice as the adipose tissue phenotype was not reported.
Lipid Synthesis
The ability to store lipids as TG is a key function of adipocytes. Impairment in TG synthesis compromises adipose tissue function and is involved in the development of lipodystrophy. AGPAT2, which is mutated in BSCL type 1 allows, the transformation of lysophosphatidic acid (LPA) to phosphatidic acid, a key step in TG synthesis. Agpat2 -/- mice lack both brown adipose tissue and WAT, and are hypertriglyceridemic, hyperinsulinemic, hyperglycemic, and severely hypoleptinemic [55]. On the basis of the severity of liver steatosis, the study authors analyzed the expression of the genes involved in hepatic lipid metabolism and found that despite AGPAT2 deficiency, lipogenesis and TG synthesis were increased in the liver of Agpat2 -/- mice. Molecular analysis revealed that the monoacylglycerol transferase 1 expression level was increased 54-fold, suggesting that the induction of the alternative monoacyl glycerol pathway might explain in part the massive liver TG accumulation in Agpat2 -/- mice. However, liver adenoviral overexpression of acylglycerol-3-phosphate-O-acyltransferase 1 (AGPAT1) and AGPAT2 in Agpat2 -/- mice failed to improve liver steatosis, suggesting that lipodystrophy and insulin resistance are critical for the liver phenotype of these animals [56•]. Indeed, cellular studies have shown that AGPAT2 (but not AGPAT1) knockdown [57] in 3T3L1 cells strongly impairs adipogenesis [58]. AGPAT2 deficiency alters lipid synthesis and leads to an accumulation of lysophosphatidic acid. Treatment with the PPARγ agonist rosiglitazone partially restored the adipogenesis [57].
Lipin 1, another key enzyme in the TG synthesis pathway, is also involved in generalized lipodystrophy in mice [59]. Spontaneous loss-of-function mutations in the gene encoding lipin 1 have been identified in the fatty liver dystrophic (Fld) mouse model, which is characterized by TG accumulation and a massive liver steatosis in the presuckling period [60]. These lipid abnormalities were associated with a strong decrease in adipose tissue LPL activity. At that time, the gene involved in the Fld phenotype was unknown. More than 10 years later, Reue et al. [59] showed that adult Fld mice have normal TG levels, only a modest liver mass increase (20 %), but a massive adipose tissue loss (80 %) associated with glucose intolerance. The adipose tissue of these mice displayed a strong morphological default with abnormal LDs. Lipin 1 exerts a phosphatidate phosphatase type 1 activity, which catalyzes the transformation of PA to diacylglycerol, also a key step in TG biosynthesis [61]. Lipin 1 deficiency strongly prevents differentiation of 3T3-L1 cells and mouse embryonic fibroblasts into adipocytes [62]. In mice, lipin 1 loss of function restricted to the mature adipocyte strongly affects TG synthesis, leads to PA accumulation, and induces lipodystrophy [63•]. Lipin 1 deficiency severely compromises adipogenesis through impairment in TG synthesis and storage in adipocytes, leading to a severe lipodystrophic phenotype. However, unlike the other lipodystrophic models, lipin 1 deficiency is not associated with hypertriglyceridemia in adult mice, in total deficient mice [59] or in adipocyte-specific deficient mice [63•]. Indeed, hypertriglyceridemia is transient and resolves after weaning, whereas lipodystrophy, in contrast, starts to be visible [59]. Lipin 1 overexpression in liver decreases VLDL secretion, whereas its deficiency increases it [64, 65]. However, liver TG synthesis is not affected in Fld mice [64], but lipin 1 has been found in the nucleus, where it amplifies the PPAR γ coactivator 1α/PPARα induction of the expression of genes involved in β-oxidation [66]. Further work is needed to understand why the effect of lipin 1 on VLDL secretion does not alter plasma TG levels in the basal condition.
Bscl2-Deficient Mouse Models: The Paradoxical Hypotriglyceridemia
BSCL2 was identified in 2001 as the first gene implicated in BSCL in humans. However, its function was unknown at that time [10]. Studies from yeast and human cells suggested that seipin is involved in LD morphology [67–70]. On the other hand, seipin deficiency strongly impairs adipogenesis [71, 72] and increases the basal rate of lipolysis, preventing, therefore, adipocyte TG accumulation [73, 74]. Seipin-deficient mice display a severe lipodystrophy associated with insulin resistance and massive liver steatosis [73–76]. Mature adipocyte AP2-driven seipin deficiency alters adipocyte function and LD morphology, and progressively leads to lipodystrophy [77]. The insights from mouse models suggest that seipin is required not only for normal adipocyte differentiation but also for the maintenance of mature adipocyte function. However, the exact function of seipin remains largely unknown. Seipin deficiency is associated with PA accumulation in yeast [78], and seipin has been shown to interact with lipin [79]. These results suggest that seipin could be involved in TG synthesis, but the exact physiological relevance of seipin/lipin interaction has not yet been established.
One striking observation, regarding the human observations, is that all three available seipin-deficient mouse models are hypotriglyceridemic in the fasting state [73–75], whereas one of the three models is also hypotriglyceridemic in the fed state [74]. We have shown that the reduced plasma TG levels could be partly due to accelerated VLDL clearance and increased TG uptake by the liver [74]. This finding raises the question of a potential liver function of seipin, in addition to its key role in adipocytes. However, thiazolidinedione (TZD) treatment improves insulin sensitivity, reduces liver steatosis, increases energy expenditure, and normalizes VLDL clearance by increasing WAT mass and adipokine (adiponectin and leptin) secretion. Nonetheless, if lipodystrophy is the key mechanism involved in the metabolic complications associated with seipin deficiency, it is quite difficult to understand why seipin-deficient mice do not display hypertriglyceridemia. It is interesting to note that the only other lipodystrophic mouse model without hypertriglyceridemia is the Fld mouse, given the fact that lipin 1 and seipin have been shown to interact.
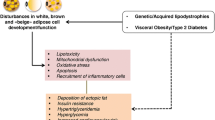
This large body of work suggests that, in general, all the genes involved play a key role in adipocyte differentiation of function, and that their deficiency leads to lipodystrophy. Most of the work that intended to show an effect of liver deletion of these genes failed to recapitulate the same phenotype. However, liver steatosis is a common feature, and the increase of lipogenesis, due to the selective hepatic insulin resistance, and the increased FFA uptake also contribute to this phenotype. Interestingly, two key parameters have not been systematically studied in these different models: VLDL production and postprandial TG clearance. When assessed, the postprandial TG clearance is delayed, which is consistent with the human kinetic studies (Fig. 1).

All the genes involved in Berardinelli–Seip congenital lipodystrophy are key for adipocyte differentiation or function. The lack of mature and functional adipocytes will basically generate three main defaults: (1) accelerated lipolysis; (2) poor lipid uptake; (3) low adipokine secretion. The inability of adipose tissue to store the dietary lipids will lead to an increased in plasma triglyceride (TG) levels, especially in the postprandial state, and an increased TG-rich lipoprotein uptake in the liver, contributing to ectopic lipid deposition. In addition, the hyperinsulinemia combined with the selective hepatic insulin resistance leads to increased de novo lipogenesis. The level of very low density lipoprotein (VLDL) secretion and its contribution to hypertriglyceridemia remain unclear. FFA free fatty acid, LD lipid droplet
Therapeutic Management of Dyslipidemias in Lipodystrophies
The main objectives in patients with lipodystrophies are to reduce hypertriglyceridemia and the cardiovascular risk. An additional aim in this population is to address NAFLD and nonalcoholic steatohepatitis (NASH), the latter being life-threatening owing to the risk of liver cirrhosis and hepatocellular carcinoma.
Apart from studies of recombinant leptin therapy, there are only very few prospective controlled studies in patients with inherited lipodystrophies, owing to the rarity of the disease. Therefore, the analysis of both efficacy and safety of available therapeutics is based on case series or expert opinion [80, 81, 102]. In some cases, we refer to studies conducted in patients with HIV-associated lipodystrophy to extrapolate some results in congenital lipodystrophies.
Lifestyle Modifications
Restriction of total fat intake to between 20 and 30 % of total dietary energy is often sufficient to maintain normal serum TG concentrations [80]. The importance of this nutritional intervention is sustained by the results obtained in Agpat2 -/- deficient mice, in which a fat-free diet for 2 weeks markedly decreased plasma and hepatic TG levels by 50 60 % [55]. In addition, high consumption of carbohydrate (especially fructose) should also be avoided. The beneficial effect of a Mediterranean diet, enriched in n − 3 polyunsaturated fatty acids (PUFAs), on metabolic parameters has been demonstrated in HIV-positive patients with highly active antiretroviral therapy (HAART)-induced metabolic syndrome [82]. Additional studies are warranted to determine whether this Mediterranean diet is equally effective in patients with inherited lipodystrophies. Finally, there are no solid data available concerning the metabolic effect of regular physical activity in patients with lipodystrophy.
n − 3 Polyunsaturated Fatty Acids
Despite the lack of controlled trials, the use of n − 3 PUFAs, eicosapentaenoic acid and docosahexaenoic acids, is recommended since they can reduce plasma TG levels by up to 50 %. Recently, n − 3 PUFAs have failed to demonstrate cardiovascular protection in a large randomized trial conducted in patients with type 2 diabetes mellitus (T2DM) or prediabetes [83].
Fibrates
Fibrates, which act as PPARα agonists, could be also used as hypotriglyceridemic drugs, although robust efficacy trials are lacking in the context of lipodystrophy [80]. Moreover, fenofibrates did not reduce the incidence of cardiovascular events in large recent randomized trials performed in patients with T2DM [84, 85].
Statins
In contrast to what observed has been observed with fibrates, statins have clearly demonstrated a consistent reduction in the incidence of cardiovascular events in several randomized trials [86]. Thus, statins should be recommended in patients with lipodystrophy, especially in those with diabetes and/or atherosclerosis. However, some pilot studies conducted in HIV patients with HAART suggest that statins are less effective in patients with lipodystrophy [87, 88]. It is unclear whether such resistance also operates in inherited lipodystrophies.
Insulin Sensitizers: Metformin and Thiazolidinediones
Since insulin resistance is one of the key feature of lipodystrophies, insulin sensitizers appear to be a logical option for the treatment of metabolic disorders.
Metformin is rather an inhibitor of hepatic gluconeogenesis than a true insulin sensitizer [89]. Metformin has been used in acquired (HAART) and congenital partial lipodystrophies. A recent meta-analysis of trials conducted in patients with HAART suggests that metformin significantly reduces both fasting insulin and plasma TG levels compared with placebo, with no effect on LDL cholesterol and HDL cholesterol. Moreover, metformin was superior to rosiglitazone in reducing lipid levels [90]. A mechanistic study of the effect of metformin (2 g/day) and rosiglitazone (8 mg/day) on postprandial lipemia has been performed in patients with HIV-associated lipodystrophy [91]. In this 6-month prospective, randomized, open study, metformin was superior to rosiglitazone for the reduction in body weight as well as in subcutaneous and abdominal fat. Metformin reduced fasting TG levels, but had no effect on the postprandial plasma TG profile. In contrast, rosiglitazone increased fasting plasma TG levels, but simultaneously decreased postprandial TG excursion, resulting in an area under the curve unchanged from the baseline. However, rosiglitazone also significantly increased the levels of remnant-like particles enriched in cholesterol, a phenomenon that might increase atherosclerosis risk [91]. No data are available for pioglitazone.
The discovery of the TZD class of antidiabetic drugs as high-affinity PPARγ ligands was a major breakthrough in the pharmacology of PPARγ. TZDs (also termed glitazones) are potent insulin sensitizers that efficiently and sustainably improve glycemic control in patients with T2DM [92]. By contrast to metformin, TZDs improve whole-body insulin sensitivity during hyperinsulinemic–euglycemic clamp studies in patients with T2DM [93, 94], reflecting an increase in peripheral glucose disposal. Although both metformin and TZDs decrease hepatic glucose production, only TZDs decrease liver fat content [93, 95]. The partitioning of lipids to WAT (i.e., “lipid steal” hypothesis) following TZD treatment likely accounts for the indirect improvement in skeletal muscle insulin sensitivity and the reduction in liver steatosis, similar to what is seen in animal models. In addition, serum adiponectin concentrations are increased by TZD treatment, but not by metformin treatment, and this increase in adiponectin levels correlates with a reduction in liver fat content [93]. However, their use has been challenged in clinical practice because of side effects which include weight gain, fluid retention precipitating cardiac failure, and bone fractures [92].
Several data sustain the efficacy of TZDs in inherited lipodystrophies. The first demonstration of the beneficial effect of TZDs came from an open-label prospective study with troglitazone in patients with various lipodystrophy syndromes [96]. After 6 months of treatment, troglitazone reduced hemoglobin A1C (HbA1C), fasting plasma TG, and FFA levels, and increased the amount of subcutaneous fat, but did not increase the amount of visceral fat [96]. The hepatoxicity observed with troglitazone was probably related to a direct toxicity of the molecule (which was withdrawn from the market shortly afterward), rather than a TZD class effect. Indeed, these seminal findings were confirmed in many individual cases reporting a decrease of fasting TG and plasma FFA levels, as well as liver enzyme levels, with either rosiglitazone treatment [97] or pioglitazone treatment [98, 99] in patients with FPL. In most of these cases, TZD was used in combination with metformin. Recently, an MRI study demonstrated that changes in adipose tissue distribution coincided with biochemical improvement in two FPL patients treated with rosiglitazone (8 mg/day). Notably, rosiglitazone induced a significant decrease (more than 50 %) in the amount of liver fat [100]. Together with the improvement of liver histological features in patients with NASH treated with pioglitazone [101], these data suggest that TZDs are the drugs of choice to treat liver damage in patients with lipodystrophy. Simha et al. [102] suggested that prolonged TZD treatment was unable to promote fat deposition in lipodystrophic regions in FPL patients, and that the increase in body weight was due to fat accumulation in nonlipodystrophic regions (i.e., truncal fat).
Compared with FPL treatment, there are only very few reports of TZD treatment in BSCL [96, 103]. As mentioned already, our data in Bscl2 -/- mice suggest a beneficial effect of TZDs in BSCL [74]. A case report sustains this hypothesis since the addition of rosiglitazone treatment (8 mg/day) to treatment with metformin (850 mg twice daily) and sulfonylurea (glimepiride) in a 19-year-old male with BSCL type 2 led to a reduction of HbA1C (-1.3 %) and fasting insulin levels and a normalization of plasma TG levels (from 4.3 to 1.7 mmol/l). Moreover, the levels of liver enzymes (alanine aminotransferase and γ-glutamyl transpeptidase) were also reduced. These metabolic effects occurred independently of changes in fat mass [103].
To precisely assess the efficacy and safety of TZDs in patients with inherited lipodystrophies, some randomized controlled trials are warranted. In the light of its beneficial effects on cardiovascular events [104], linked to a better plasma lipid profile than rosiglitazone [105], as well as its positive action on NASH [101], pioglitazone should be chosen first.
Recombinant Leptin Replacement Therapy
Marked improvements in metabolic parameters were observed with leptin replacement therapy in patients with generalized lipodystrophies. Recently, the long-term (i.e., 3 years) effects of metreleptin (an analogue of human leptin) on dyslipidemia were reported in 55 patients with lipodystrophy (36 BSCL patients and 19 partial lipodystrophy patients) [106]. Notably, the mean percentage of TG reduction was 36.7 % after 4 months of treatment and 35.4 % at 3 years. The reduction was even more drastic in patients with elevated plasma TG levels at the baseline (200 mg/dl or higher), with a 51.2 % decrease at 3 years. In addition, metreleptin also reduced total cholesterol and LDL levels (-105.8 % and -64.3 % at 3 years, respectively), whereas there was no effect on HDL levels. Leptin recombinant therapy also exerts some beneficial action on glucose homeostasis, with a reduction of the levels of HbA1C and liver enzymes (suggesting a beneficial effect on NAFLD). In some patients, a resistance to leptin replacement therapy has been observed, linked to the development of antibodies against recombinant leptin [107]. In a 28-month trial conducted in eight children with BSCL, a negative or a partial effect of leptin therapy on hypertriglyceridemia, liver steatosis, and insulin resistance was observed in five patients, despite a significant increase in leptin dosage. This lack of efficacy was related to neutralizing leptin antibodies.
Conclusion
Adipose tissue failure leads to numerous metabolic complications. The commonet lipid disorder trait associated with lipodystrophy is hypertriglyceridemia. Uncontrolled lipolysis and the inability of adipocytes to store dietary lipids will promote TG and FFA accumulation in plasma. Increased lipid liver uptake combined with increased lipogenesis contributes to liver steatosis. Animal models are unique tools to dissect the molecular mechanism involved in plasma TG regulation in the context of lipodystrophy. Notably, the respective dietary lipogenesis versus de novo liver lipogenesis has not been established yet.
Interestingly, most of the mechanistic studies suggest that the adipocyte function of the different genes involved, rather that their liver function, is at the molecular basis of the lipid disorder associated with lipodystrophy. However, the unexpected hypotriglyceridemic phenotype of Bscl2 -/- mice as well as the normal TG levels of adult Fld mice suggest that, at least in mice, the adipose failures which result from different causes do not have similar consequences for circulating TG.
The use of animal models is also a unique opportunity to identify new targets and to test new therapeutic approaches. As tested in mice 15 years ago, leptin therapy in humans has beneficial effects but remains a very heavy severe treatment, with the risk of developing antibodies. Case reports and animals studies suggest that pioglitazone might be beneficial to improve both lipodystrophy and the associated metabolic complications in BSCL. Clinical studies with kinetic lipid assessment will be needed to establish if TZDs are appropriate in the clinical management of lipodystrophy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Garg A. Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96:3313–25. This is a very comprehensive review in terms of clinical classification of the different lipodystrophic syndromes.
Huang-Doran I, Sleigh A, Rochford JJ, O’Rahilly S, Savage DB. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol. 2010;207:245–55.
Vatier C, Bidault G, Briand N, Guénantin AC, Teyssières L, Lascols O, et al. What the genetics of lipodystrophy can teach us about insulin resistance and diabetes. Curr Diabetes Rep. 2013;13:757–67.
Vantyghem MC, Balavoine AS, Douillard C, Defrance F, Dieudonne L, Mouton F, et al. How to diagnose a lipodystrophy syndrome. Ann Endocrinol (Paris). 2012;73:170–89.
Seip M, Trygstad O. Generalized lipodystrophy, congenital and acquired (lipoatrophy). Acta Paediatr Suppl. 1996;413:2–28.
Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87:2395–8.
Antuna-Puente B, Boutet E, Vigouroux C, Lascols O, Slama L, Caron-Debarle M, et al. Higher adiponectin levels in patients with Berardinelli-Seip congenital lipodystrophy due to seipin as compared with 1-acylglycerol-3-phosphate-O-acyltransferase-2 deficiency. J Clin Endocrinol Metab. 2010;95:1463–8.
Garg A, Wilson R, Barnes R, Arioglu E, Zaidi Z, Gurakan F, et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab. 1999;84:3390–4.
Agarwal AK, Arioglu E, De Almeida S, Akkoc N, Taylor SI, Bowcock AM, et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet. 2002;31:21–3.
Magré J, Delépine M, Khallouf E, Gedde-Dahl TJ, Van Maldergem L, Sobel E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28:365–70.
Magré J, Delépine M, Van Maldergem L, Robert JJ, Maassen JA, Meier M, et al. Prevalence of mutations in AGPAT2 among human lipodystrophies. Diabetes. 2003;52:1573–8.
Kim CA, Delépine M, Boutet E, El Mourabit H, Le Lay S, Meier M, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93:1129–34.
Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet. 2010;6(3):e1000874.
Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119:2623–33.
Knebel B, Kotzka J, Lehr S, Hartwig S, Avci H, Jacob S, et al. A mutation in the c-fos gene associated with congenital generalized lipodystrophy. Orphanet J Rare Dis. 2013;8:119.
Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–34.
Peters JM, Barnes R, Bennett L, Gitomer WM, Bowcock AM, Garg A. Localization of the gene for familial partial lipodystrophy (Dunnigan variety) to chromosome 1q21-22. Nat Genet. 1998;18:292–5.
Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24:153–6.
Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9:109–12.
Speckman RA, Garg A, Du F, Bennett L, Veile R, Arioglu E, et al. Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet. 2000;66:1192–8.
Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, et al. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–3.
George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–8.
Rubio-Cabezas O, Puri V, Murano I, Saudek V, Semple RK, Dash S, et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1:280–7.
Gandotra S, Le Dour C, Bottomley W, Cervera P, Giral P, Reznik Y, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364:740–8.
Van Maldergem L, Magré J, Gedde-DahlJr T, Khallouf E, Delépine M, Trygstad O, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39:722–33.
Adiels M, Taskinen MR, Packard C, Caslake MJ, Soro-Paavonen A, Westerbacka J, et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia. 2006;49:755–65.
Beylot M, Sautot G, Laville M, Cohen R. Metabolic studies in lipoatrophic diabetes: mechanism of hyperglycemia and evidence of resistance to insulin of lipid metabolism. Diabete Metab. 1988;14:20–4.
Wajchenberg BL, Amâncio RF, Santomauro AT, Maranhão RC. Metabolism of chylomicrons in patients with congenital lipoatrophic diabetes: a study with emulsion models of chylomicrons. Clin Endocrinol (Oxf). 2004;61:347–52.
Kodama S, Kasuga M, Seki A, Ninomiya M, Sakurai T, Morishita Y, et al. Congenital generalized lipodystrophy with insulin-resistant diabetes. Eur J Pediatr. 1978;127:111–9.
Naito C, Togawa K. A possible role of circulating lipoprotein-triglycerides in the increase in concentration of free fatty acids and in insulin resistance in “total” lipodystrophy. J Clin Endocrinol Metab. 1974;39:1030–7.
Enzi G, Digito M, Baldo-Enzi G, Cominacini L, Dodi G, Carraro R, et al. Lipid metabolism in lipoatrophic diabetes. Horm Metab Res. 1988;20:587–91.
Tan GD, Savage DB, Fielding BA, Collins J, Hodson L, Humphreys SM, et al. Fatty acid metabolism in patients with PPARγ mutations. J Clin Endocrinol Metab. 2008;93:4462–70.
Savage DB, Murgatroyd PR, Chatterjee VK, O’Rahilly S. Energy expenditure and adaptive responses to an acute hypercaloric fat load in humans with lipodystrophy. J Clin Endocrinol Metab. 2005;90:1446–52.
Savage DB, Semple RK, Clatworthy MR, Lyons PA, Morgan BP, Cochran EK, et al. Complement abnormalities in acquired lipodystrophy revisited. J Clin Endocrinol Metab. 2009;94:10–6.
Ross R, Graves A, Spiegelman M. Targeted expression of a toxin gene to adipose tissue: transgenic mice resistant to obesity. Genes Dev. 1993;7:1318–24.
Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL, et al. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev. 1998;12:3182–94.
Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, et al. Life without white fat: a transgenic mouse. Genes Dev. 1998;12:3168–81.
Linhart HG, Ishimura-Oka K, DeMayo F, Kibe T, Repka D, Poindexter B, et al. C/EBPα is required for differentiation of white, but not brown, adipose tissue. Proc Natl Acad Sci U S A. 2001;98:12532–7.
Gavrilova O, Marcus-Samuels B, Graham D, Kim JK, Shulman GI, Castle AL, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105:271–8.
Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401:73–6.
Ebihara K, Ogawa Y, Masuzaki H, Shintani M, Miyanaga F, Aizawa-Abe M, et al. Transgenic overexpression of leptin rescues insulin resistance and diabetes in a mouse model of lipoatrophic diabetes. Diabetes. 2001;50:1440–8.
Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, et al. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–7.
Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, et al. PPARγ is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–95.
He W, Barak Y, Hevener A, Olson P, Liao D, Le J, et al. Adipose-specific peroxisome proliferator-activated receptor γ knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A. 2003;100:15712–7.
Wang F, Mullican SE, DiSpirito JR, Peed LC, Lazar MA. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARγ. Proc Natl Acad Sci U S A. 2013;110:18656–61. Complete deletion of PPARγ in mature adipocytes has been quite a challenge during the last 10 years. This article provides important insight into the role of PPARγ after the initiation of adipogenesis.
Razani B, Combs TP, Wang XB, Frank PG, Park DS, Russell RG, et al. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem. 2002;277:8635–47.
Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–82.
Cohen AW, Razani B, Wang XB, Combs TP, Williams TM, Scherer PE, et al. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol. 2003;285:C222–35.
Le Lay S, Blouin CM, Hajduch E, Dugail I. Filling up adipocytes with lipids. Lessons from caveolin-1 deficiency. Biochim Biophys Acta. 2009;1791:514–8.
Le Lay S, Hajduch E, Lindsay MR, Le Liepvre X, Thiele C, Ferre P, et al. Cholesterol-induced caveolin targeting to lipid droplets in adipocytes: a role for caveolar endocytosis. Traffic. 2006;7:549–61.
Blouin CM, Le Lay S, Eberl A, Kofeler HC, Guerrera IC, Klein C, et al. Lipid droplet analysis in caveolin-deficient adipocytes: alterations in surface phospholipid composition and maturation defects. J Lipid Res. 2010;51:945–56.
Le Lay S, Briand N, Blouin CM, Chateau D, Prado C, Lasnier F, et al. The lipoatrophic caveolin-1 deficient mouse model reveals autophagy in mature adipocytes. Autophagy. 2010;6:754–63.
Martin S, Fernandez-Rojo MA, Stanley AC, Bastiani M, Okano S, Nixon SJ, et al. Caveolin-1 deficiency leads to increased susceptibility to cell death and fibrosis in white adipose tissue: characterization of a lipodystrophic model. PLoS One. 2012;7:e46242.
Frank PG, Lee H, Park DS, Tandon NN, Scherer PE, Lisanti MP. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:98–105.
Cortés VA, Curtis DE, Sukumaran S, Shao X, Parameswara V, Rashid S, et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab. 2009;9:165–76.
Agarwal AK, Sukumaran S, Cortés VA, Tunison K, Mizrachi D, Sankella S, et al. Human 1-acylglycerol-3-phosphate O-acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2-/- gene lipodystrophic mice. J Biol Chem. 2011;286:37676–91. In this study, the authors demontrate that liver re-expression of AGPAT2 is not sufficient to rescue the severe liver steatosis phenotype.
Subauste AR, Das AK, Li X, Elliott BG, Elliot B, Evans C, et al. Alterations in lipid signaling underlie lipodystrophy secondary to AGPAT2 mutations. Diabetes. 2012;61:2922–31.
Gale SE, Frolov A, Han X, Bickel PE, Cao L, Bowcock A, et al. A regulatory role for 1-acylglycerol-3-phosphate-O-acyltransferase 2 in adipocyte differentiation. J Biol Chem. 2006;281:11082–9.
Reue K, Xu P, Wang XP, Slavin BG. Adipose tissue deficiency, glucose intolerance, and increased atherosclerosis result from mutation in the mouse fatty liver dystrophy (fld) gene. J Lipid Res. 2000;41:1067–76.
Langner CA, Birkenmeier EH, Ben-Zeev O, Schotz MC, Sweet HO, Davisson MT, et al. The fatty liver dystrophy (fld) mutation. A new mutant mouse with a developmental abnormality in triglyceride metabolism and associated tissue-specific defects in lipoprotein lipase and hepatic lipase activities. J Biol Chem. 1989;264:7994–8003.
Donkor J, Sariahmetoglu M, Dewald J, Brindley DN, Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J Biol Chem. 2007;282:3450–7.
Phan J, Péterfy M, Reue K. Lipin expression preceding peroxisome proliferator-activated receptor-γ is critical for adipogenesis in vivo and in vitro. J Biol Chem. 2004;279:29558–64.
Nadra K, Médard JJ, Mul JD, Han GS, Grès S, Pende M, et al. Cell autonomous lipin 1 function is essential for development and maintenance of white and brown adipose tissue. Mol Cell Biol. 2012;32:4794–810. In this study, the authors demonstrate for the first time that adipocyte-specific deletion of lipin 1 is responsible for the lipodystrophic phenotype in Fld mice. This is a very complete invivo and invitro study.
Chen Z, Gropler MC, Norris J, Lawrence JC, Harris TE, Finck BN. Alterations in hepatic metabolism in fld mice reveal a role for lipin 1 in regulating VLDL-triacylglyceride secretion. Arterioscler Thromb Vasc Biol. 2008;28:1738–44.
Hu M, Yin H, Mitra MS, Liang X, Ajmo JM, Nadra K, et al. Hepatic-specific lipin-1 deficiency exacerbates experimental alcohol-induced steatohepatitis in mice. Hepatology. 2013;58:1953–63.
Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–22.
Fei W, Shui G, Gaeta B, Du X, Kuerschner L, Li P, et al. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J Cell Biol. 2008;180:473–82.
Szymanski KM, Binns D, Bartz R, Grishin NV, Li WP, Agarwal AK, et al. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci U S A. 2007;104:20890–5.
Boutet E, El Mourabit H, Prot M, Nemani M, Khallouf E, Colard O, et al. Seipin deficiency alters fatty acid Δ9 desaturation and lipid droplet formation in Berardinelli-Seip congenital lipodystrophy. Biochimie. 2009;91:796–803.
Cartwright BR, Goodman JM. Seipin: from human disease to molecular mechanism. J Lipid Res. 2012;53:1042–55.
Payne VA, Grimsey N, Tuthill A, Virtue S, Gray SL, Dalla Nora E, et al. The human lipodystrophy gene BSCL2/seipin may be essential for normal adipocyte differentiation. Diabetes. 2008;57:2055–60.
Chen W, Yechoor VK, Chang BH, Li MV, March KL, Chan L. The human lipodystrophy gene product Berardinelli-Seip congenital lipodystrophy 2/seipin plays a key role in adipocyte differentiation. Endocrinology. 2009;150:4552–61.
Chen W, Chang B, Saha P, Hartig SM, Li L, Reddy VT, et al. Berardinelli-Seip congenital lipodystrophy 2/seipin is a cell-autonomous regulator of lipolysis essential for adipocyte differentiation. Mol Cell Biol. 2012;32:1099–111.
Prieur X, Dollet L, Takahashi M, Nemani M, Pillot B, Le May C, et al. Thiazolidinediones partially reverse the metabolic disturbances observed in Bscl2/seipin-deficient mice. Diabetologia. 2013;56:1813–25.
Cui X, Wang Y, Tang Y, Liu Y, Zhao L, Deng J, et al. Seipin ablation in mice results in severe generalized lipodystrophy. Hum Mol Genet. 2011;20:3022–30.
Dollet L, Magré J, Cariou B, Prieur X. Function of seipin: new insights from Bscl2/seipin knockout mouse models. Biochimie. 2014;96:166–72.
Liu L, Jiang Q, Wang X, Zhang Y, Lin RC, Lam SM, et al. Adipose-specific knockout of Seipin/Bscl2 results in progressive lipodystrophy. Diabetes. 2014. doi:10.2337/db13-0729.
Fei W, Shui G, Zhang Y, Krahmer N, Ferguson C, Kapterian TS, et al. A role for phosphatidic acid in the formation of “supersized” lipid droplets. PLoS Genet. 2011;7:e1002201.
Sim MF, Dennis RJ, Aubry EM, Ramanathan N, Sembongi H, Saudek V, et al. The human lipodystrophy protein seipin is an ER membrane adaptor for the adipogenic PA phosphatase lipin 1. Mol Metab. 2012;2:38–46.
Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine (Baltimore). 2003;82:129–46.
Handelsman Y, Oral EA, Bloomgarden ZT, Brown RJ, Chan JL, Einhorn D, et al. The clinical approach to the detection of lipodystrophy - an AACE consensus statement. Endocr Pract. 2013;19:107–16.
Tsiodras S, Poulia KA, Yannakoulia M, Chimienti SN, Wadhwa S, Karchmer AW, et al. Adherence to Mediterranean diet is favorably associated with metabolic parameters in HIV-positive patients with the highly active antiretroviral therapy-induced metabolic syndrome and lipodystrophy. Metabolism. 2009;58:854–9.
Bosch J, Gerstein HC, Dagenais GR, Díaz R, Dyal L, Jung H, et al. n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med. 2012;367:309–18.
Ginsberg HN, Elam MB, Lovato LC, Crouse JR, Leiter LA, Linz P, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74.
Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–61.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78.
Johns KW, Bennett MT, Bondy GP. Are HIV positive patients resistant to statin therapy? Lipids Health Dis. 2007;6:27.
Mallon PW, Miller J, Kovacic JC, Kent-Hughes J, Norris R, Samaras K, et al. Effect of pravastatin on body composition and markers of cardiovascular disease in HIV-infected men–a randomized, placebo-controlled study. AIDS. 2006;20:1003–10.
Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122:253–70.
Sheth SH, Larson RJ. The efficacy and safety of insulin-sensitizing drugs in HIV-associated lipodystrophy syndrome: a meta-analysis of randomized trials. BMC Infect Dis. 2010;10:183.
van Wijk JP, Hoepelman AI, de Koning EJ, Dallinga-Thie G, Rabelink TJ, Cabezas MC. Differential effects of rosiglitazone and metformin on postprandial lipemia in patients with HIV-lipodystrophy. Arterioscler Thromb Vasc Biol. 2011;31:228–33.
Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends Endocrinol Metab. 2012;23:205–15.
Tiikkainen M, Häkkinen AM, Korsheninnikova E, Nyman T, Mäkimattila S, Yki-Järvinen H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes. 2004;53:2169–76.
Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, et al. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2002;87:2784–91.
Juurinen L, Kotronen A, Granér M, Yki-Järvinen H. Rosiglitazone reduces liver fat and insulin requirements and improves hepatic insulin sensitivity and glycemic control in patients with type 2 diabetes requiring high insulin doses. J Clin Endocrinol Metab. 2008;93:118–24.
Arioglu E, Duncan-Morin J, Sebring N, Rother KI, Gottlieb N, Lieberman J, et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann Intern Med. 2000;133:263–74.
Lüdtke A, Heck K, Genschel J, Mehnert H, Spuler S, Worman HJ, et al. Long-term treatment experience in a subject with Dunnigan-type familial partial lipodystrophy: efficacy of rosiglitazone. Diabet Med. 2005;22:1611–3.
Gambineri A, Semple RK, Forlani G, Genghini S, Grassi I, Hyden CS, et al. Monogenic polycystic ovary syndrome due to a mutation in the lamin A/C gene is sensitive to thiazolidinediones but not to metformin. Eur J Endocrinol. 2008;159:347–53.
Moreau F, Boullu-Sanchis S, Vigouroux C, Lucescu C, Lascols O, Sapin R, et al. Efficacy of pioglitazone in familial partial lipodystrophy of the Dunnigan type: a case report. Diabetes Metab. 2007;33:385–9.
McLaughlin PD, Ryan J, Hodnett PA, O’Halloran D, Maher MM. Quantitative whole-body MRI in familial partial lipodystrophy type 2: changes in adipose tissue distribution coincide with biochemical improvement. AJR Am J Roentgenol. 2012;199:W602–6.
Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–307.
Simha V, Rao S, Garg A. Prolonged thiazolidinedione therapy does not reverse fat loss in patients with familial partial lipodystrophy, Dunnigan variety. Diabetes Obes Metab. 2008;10:1275–6.
Victoria B, Cabezas-Agricola JM, Gonzalez-Mendez B, Lattanzi G, Del Coco R, Loidi L, et al. Reduced adipogenic gene expression in fibroblasts from a patient with type 2 congenital generalized lipodystrophy. Diabet Med. 2010;27:1178–87.
Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, et al. A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care. 2005;28:1547–54.
Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–89.
Chan JL, Lutz K, Cochran E, Huang W, Peters Y, Weyer C, et al. Clinical effects of long-term metreleptin treatment in patients with lipodystrophy. Endocr Pract. 2011;17:922–32.
Beltrand J, Lahlou N, Le Charpentier T, Sebag G, Leka S, Polak M, et al. Resistance to leptin-replacement therapy in Berardinelli-Seip congenital lipodystrophy: an immunological origin. Eur J Endocrinol. 2010;162:1083–91.
Acknowledgments
This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale (Inserm), the French Ministère de l’Enseignement Supérieur et de la Recherche, and French associations (Aide aux Jeunes Diabétiques, Fondation de France, Fondation GenaVie, Association de Langue Française pour l’Etude du Diabète et des Maladies Métaboliques/Société Francophone du Diabète, and Assocation pour la Recherche Diabète).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Xavier Prieur, Cedric Le May, Jocelyne Magré, and Bertrand Cariou declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Rare Diseases and Lipid Metabolism
Rights and permissions
About this article

Cite this article
Prieur, X., Le May, C., Magré, J. et al. Congenital Lipodystrophies and Dyslipidemias. Curr Atheroscler Rep 16, 437 (2014). https://doi.org/10.1007/s11883-014-0437-x
Published:
DOI: https://doi.org/10.1007/s11883-014-0437-x