
Abstract
The concept of the vulnerable atherosclerotic plaque first developed through histological evaluation of post-mortem coronary arteries has been significantly advanced in recent years by new imaging modalities. Imaging has: 1) verified histological findings, 2) identified features that are associated with unstable plaque, 3) followed plaques over time to study the dynamic nature of vulnerable plaque, 4) predicted clinical events based on imaging features, 5) tested the impact of medical interventions on plaque morphology. This review will summarize the major findings of imaging studies with a focus on how the knowledge base of vulnerable plaque has been advanced.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
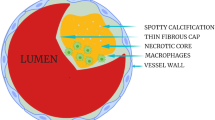
Coronary artery disease accounts for nearly 500,000 deaths and 1,000,000 myocardial infarctions per year in the US [1]. The sudden reduction of blood flow to the myocardium leads to myocardial infarction (MI), chest pain, lethal arrhythmias, heart failure, and cardiogenic shock [2]. Such acute coronary syndromes (ACS) generally result from the formation of a thrombus overlying a ruptured thin cap fibroatheroma (TCFA, 65 % of cases), over an erosion of a proteoglycan rich lesion (35 % of cases) or occasionally over a calcified nodule [3]. By and large, research has centered on the development and causes of TCFA instability by studying the manner in which fibroatheromas (either thin or thick cap) can progress and subsequently rupture or regress to a more stable phenotype.
Utilizing recursive partitioning analysis, Narula et. al showed that a fibrous cap thickness of <55 μm was the best discriminator of plaque instability causing sudden death. Vulnerable plaques also had larger necrotic cores and greater macrophage infiltration [4•]. Computer modeling, however, has shown that the degree of vascular remodeling and necrotic core size, in addition to fibrous cap thickness, play a role in the development of unstable lesions [5].
While autopsy studies have established the composition and structure of high-risk lesions, there are inherent limitations. The presence of plaque rupture with thrombus does not prove causality [3] and sudden cardiac death occurs in the absence of plaque rupture [6]. By definition, these studies are biased towards those clinical events that caused cardiac death and not the less significant clinical events caused by plaque instability. Finally, autopsy studies cannot render information on lesion progression, the timeline of high risk plaque development, predictive features of future vulnerable plaques, or the impact of medical therapy on plaque progression and regression. Recent results using novel non-invasive and invasive imaging modalities possessing the ability to define important features of plaque composition and structure has lent insight into the dynamic nature of atherosclerotic plaques. Table 1 summarizes the main features of the imaging modalities covered in this review.
Non-Invasive Imaging Systems
Computed Tomography Angiography
Contrast enhanced coronary computed tomography angiography (CTA) is typically performed with a 64-slice multi-detector scanner after the injection of iodinated contrast. The contrast enhances the arterial lumen. Calcium is visualized as high attenuation material while soft non-calcified plaque has a lower attenuation, so plaques are categorized as non-calcified, partially calcified or calcified. CTA can determine the degree of stenosis as well as such atherosclerotic changes as plaque volume, positive vascular remodeling, lipoprotein deposition (non-calcified plaques), and calcification [7]. CTA is limited by a spatial resolution of about 400 μm.
Positron Emission Tomography with 18 F-Fluorodeoxyglucose
Positron emission tomography (PET) with 18 F-fluorodeoxyglucose (FDG) measures metabolic activity by assessing glucose utilization, thereby detecting active plaque inflammation, a sign of vulnerable lesions. 18 F-FDG, a glucose analog with the positron-emitting radioactive isotope fluorine-18, is injected intravenously and is taken up by cells that utilize glucose for metabolic activity. As the 18 F-FDG undergoes radioactive decay, a PET camera detects and localizes emitted gamma rays. In atherosclerotic arteries, activated macrophages with up to 50-fold higher glycolytic activity than background tissue take up 18 F-FDG and can be visualized as areas of active inflammation. The spatial resolution of active inflammation is significantly improved by combining PET/FDG with CT [8]. However, respiratory and cardiac motion during the cardiac cycle combined with poor spatial resolution make identification of the specific location of plaque inflammation difficult and high glucose utilization of the myocardium creates substantial background noise limiting this technology [8, 9].
Invasive Imaging Techniques
Intravascular Ultrasonography
Intravascular ultrasound (IVUS) imaging systems consist of a catheter incorporating a miniaturized ultrasound transducer of either a single piezoelectric element with rotating mirror or an array of multiple piezoelectric elements. The ultrasound frequencies are 30 – 50 MHz yielding a practical axial resolution of about 150 – 250 μm and a lateral resolution of 250 μm, limiting assessment of fibrous cap thickness. Ultrasound waves are reflected at the interface of materials which have significant differences in acoustic impendence such as between blood and the leading edge of the intimal layer and the external elastic lamina. The medial layer is sonolucent and the interface between the intima and media is not well visualized secondary to similar acoustic impedance, hence plaque burden is read out as the sum of the intimal and medial areas. Virtual histology intravascular ultrasound (VH-IVUS) uses advanced processing of grayscale IVUS signals by employing spectral analysis of back-scatter signals. This allows such plaque features as necrotic core, calcium, fibrous tissue and lipid core to be better visualized [9]. VH-IVUS has the same resolution as IVUS.
Optical Coherence Tomography
Optical coherence tomography (OCT) measures the echo time delay and intensity of light that is reflected or back-scattered from internal structures in tissue to produce cross-sectional images [10]. Because OCT uses near-infrared light, a higher axial spatial resolution of 10 – 20 μm is achieved [9]. OCT visualizes all three arterial layers and can accurately assess high risk plaque features such as fibrous cap thickness, necrotic core, lipid content, calcium, nodules, plaque rupture, dissection, erosions, and small platelet-rich and thrombin-rich thrombi [10]. Early ex vivo studies on atherosclerotic arterial segments showed that OCT has a high sensitivity and specificity for discriminating between fibrous, fibrocalcific and lipid-rich plaques [11] and may be able to identify macrophage infiltration of the fibrous cap, a key characteristic of vulnerable plaques [12]. OCT is limited by a penetration depth between 0.5 and 1.5 mm, making assessment of larger arteries difficult [10].
Near-Infrared Spectroscopy
Near-infrared spectroscopy (NIRS), designed to quantify lipid core, emits infrared light to the vessel wall and collects absorbable spectra over a wide range of wavelengths. By using a scanning near-infrared laser, the catheter can acquire 32,000 chemical measurements per 100 mm of artery at a pullback rate of 0.5 mm/s with several millimeters of penetration into tissue. Lipid core cholesterol has a unique spectral identification allowing the NIRS catheter to detect and quantify the amount of lipid in a plaque. Since NIRS only provides compositional data, a combination NIRS/IVUS catheter has been designed to provide both plaque characterization as well as chemical composition [13] (Fig. 1).

NIRS/IVUS representative image. IVUS/NIRS obtained from the culprit lesion causing a NSTEMI in a 63 year old woman. Image on the left demonstrates a high-grade lesion (area between dashed and dotted circles) with attenuated plaque between 3 and 10 o’clock. The yellow spectral circle on the outside displays the NIRS findings of the plaque showing that the area between 5 and 10 o’clock is a lipid core plaque. The top right display represents the lipid signal in yellow along the longitudinal length of the artery while the bottom right display integrates the longitudinal IVUS data with the NIRS chemogram. The green vertical line designates the lesion location along the artery. The dashed circle represents the external elastic lamina and the dotted circle represents the lumen lesion interface
Angioscopy
Angioscopy, a fiber-optic bundle mounted on a guidewire, directly visualizes the arterial luminal surface. Atherosclerotic plaques are seen as well-demarcated white, yellow or orange protrusions into the lumen. Deep yellow and yellow-red lesions denote lipid-rich atheromas with necrotic cores and a thin fibrous cap, respectively, while gray-white color corresponds with fibrous plaques. Patients with unstable angina generally have predominantly yellow lesions, commonly with ruptures and red thrombi [14]. A higher intensity of angioscopic yellow color was associated with thinner fibrous caps by OCT [15].
Identifying High-Risk Lesions
Imaging studies have corroborated necropsy findings by associating vulnerable characteristics of lesions in vivo with clinical events. Specifically plaques with a large plaque burden, a high lipid content, and a thinner inflamed fibrous cap have generally been shown to be associated with coronary events. However, lesions other than TCFAs have also been observed to cause thrombosis and coronary events.
Although coronary angiography is limited by the ability to visualize only the arterial lumen and not the wall, early angiography studies introduced the important concept of vulnerable plaque. These studies showed that patients harbor angiographically mild non-culprit lesions with the potential to cause recurrent ischemic events. Ambrose et.al, in 1988 [16], demonstrated that lesions causing subsequent myocardial infarctions were not initially angiographically severe. In 23 patients who had an initial cardiac catheterization and then went on to develop an MI, the median percent stenosis was 48 %, and the patients with q wave infarctions had less severe initial stenoses than patients with non-q wave infarctions (34 % vs. 80 % stenosis). Cutlip et. al followed 1228 patients for 5 years and showed a 1-year 12.4 % event rate and a subsequent 6.3 %/year event rate of non-culprit lesions in patients undergoing culprit lesion stent implantation [17]. A study of 3,747 patients undergoing PCI showed that 10.8 % of patients required revascularization of non-culprit lesions within a year for newly developed symptoms with 68.8 % presenting with an ACS. The initial severity of lesions requiring subsequent revascularization was 41.8 ± 20.8 % [18].
While angiographic studies have shown that lesions which later progressed to cause clinical events generally were angiographically mild in severity, CTA and/or IVUS demonstrated that plaque burden is an important determinate of future instability. By CTA, culprit lesions in patients with ACS have significantly greater plaque area in addition to a higher remodeling index, higher prevalence of non-calcific plaque, evidence of spotty calcification and contrast rims, and lower radiographic density [19–21]. In an IVUS study of 74 ACS patients comparing culprit with non-culprit lesions, culprit lesions had smaller lumen areas and greater plaque burden [22]. The plaque composition has also been shown to be associated with clinical events. By NIRS, culprit lesions in ACS patients were more likely to have a high lipid core compared to non-culprit lesions (84.4 % vs. 52.8 %, p = 0.004), and non-culprit lesions in ACS patients were more likely to have a high lipid core compared to stable angina patients [23•]. In another NIRS study of 20 patients presenting with STEMI comparing culprit to non-culprit lesions and to autopsy control segments, the maximum lipid core burden index distinguished culprit lesions from non-culprit lesions. This index value was nearly six times greater in culprit lesions and 87 times greater than autopsy segments free of large lipid core plaque. A value of > 400 had a sensitivity of 85 % and a specificity of 98 % for distinguishing between culprit and non-culprit lesions [24].
Lesions with thin fibrous caps have been shown to correlate strongly with coronary events by multiple imaging studies. Jang et. Al, using OCT, analyzed lesions causing a recent acute MI, ACS or stable angina and showed that plaques with thinner fibrous caps were more common in MI and ACS patients compared to stable angina patients. The median minimum thickness of the fibrous cap was 47.0, 53.8, and 102.6 μm in the MI, ACS, and stable angina groups, respectively (P < 0.034). The frequency of TCFAs (fibrous cap thickness of <65 μm) was 72 % in the MI, 50 % in the ACS, and 20 % in the stable angina group (P = 0.012) [25]. Kubo et. al compared culprit lesions in MI and stable angina patients and showed increased plaque rupture and intracoronary thrombus and significantly thinner fibrous caps in MI lesions. Non-culprit TCFAs also were significantly more common in MI patients [26]. By comparing cap thickness by OCT in 71 ruptured plaques and 111 non-ruptured plaques, ruptured plaque median thinnest cap thickness was 54 μm (range 50 – 60 μm) and the median most representative cap thickness was 116 μm (range 103 – 136 μm). For non-ruptured plaques, the results were 80 μm (67 – 104 μm) and 182 μm (156 – 216 μm), respectively. The best cut-offs for predicting rupture were <67 μm for the thinnest cap thickness and <151 μm for the most representative cap thickness [27]. Of interest was a study showing that ACS patients with symptoms at rest have rupture of thinner caps than those that rupture with exertion (50 μm vs. 90 μm), evidence that there may be a spectrum of plaque vulnerability and rupture [28].
While a fibrous cap thickness of <65 μm is a strong predictor of subsequent plaque rupture, two studies have shown that ruptures also occur in fibroatheromas with thicker fibrous caps. By OCT, Tanaka et. al showed that 33 % of ruptured plaques in acute coronary syndrome patients possessed a fibrous cap thickness of >65 μm up to 140 μm in thickness [28]. In STEMI patients, Toutouzas et. al also demonstrated by OCT that fibrous cap thickness ranged from 30 – 140 μm. In only 50.9 % of patients did an OCT-defined TCFA cause the infarct [29•]. Ino et. al showed a higher incidence of plaque rupture (70 % vs. 47 %, P = 0.33), thin-cap fibroatheroma (78 % vs. 49 %, P = 0.008) and red thrombus (78 % vs. 27 %, P < 0.001) by OCT in culprit lesions causing STEMI compared to lesions causing non-ST elevation ACS. In addition, while the lumen area at the site was similar in both groups, the ruptured cavity area was significantly larger in the lesions causing STEMI [30].
Imaging studies have also identified unique plaque characteristics that correlate with clinical events. By OCT, plaques of ACS patients had in addition to thinner fibrous caps, a wider lipid arc, larger lipid volume, and increased number of macrophages compared to non-ACS patients [31]. By VH-IVUS, ACS patients had more lesions containing plaque ruptures and thrombosis and VH-TCFAs with larger necrotic cores and less fibrous tissue compared to stable angina patients [32]. Certain calcium patterns including a larger number of calcium deposits but within an arc of less than 90 degrees have been associated with plaque rupture [33]. Correlating findings with OCT or VH-IVUS, CTA identified TCFAs on the basis of positive remodeling, low plaque attenuation, and ring-like enhancement [34, 35]. A napkin-ring sign by CTA, a ring-like attenuation of a non-calcified plaque, indicated the presence of a high-risk lesion with 98.9 % specificity compared to histology (95 % CI: 97.6 % to 100 %) [36•]. This finding also had an 88 % positive predictive value for detecting disrupted plaque identified by angioscopy [37]. Plaque neovascularization can only be identified by OCT as micro-channels and correlated with plaque rupture, thinner fibrous caps, and elevated levels of hs-CRP [38] and lesion progression [39]. Finally, imaging studies that detect inflammation have shown the importance of the increased presence of macrophages in unstable plaques. By OCT, plaque rupture sites demonstrated significantly greater macrophage density [40] and in an FDG-PET study comparing 10 patients with ACS to 15 patients with stable angina, the ACS patients had higher FDG uptake in the ascending aorta and left main coronary artery. Culprit lesions had greater FDG uptake than non-culprit lesions demonstrating on-going plaque inflammation in vulnerable lesions [41].
While a high plaque burden and a thin fibrous cap are unstable features that define a TCFA, other studies have identified lesion anatomic categories other than TCFA that may cause acute vessel thrombosis. Kim et. al, using VH-IVUS to assess the incidence of culprit plaque rupture in 172 patients with ST-elevation MI showed that only half the plaque ruptures demonstrated findings of a TCFA, i.e. plaque burden over 40 % and necrotic core over 10 % of plaque area, while the other half showed features not thought to be indicate vulnerability. These included 1 % thick capped fibroatheromas, 19 % fibro-calcified plaques and 11 % showing pathologic intimal thickening [42•]. Multi-modality imaging using both VH-IVUS and angioscopy in patients presenting with ACS showed that 54.8 % of thrombotic lesions had no rupture [43]. Analysis by VH-IVUS showed that thrombotic lesions with and without plaque rupture were very similar, except that thrombotic lesions without plaque rupture were more likely to be a TCFA (74 % vs. 58 %) with a significantly larger necrotic core. These data suggest other pathophysiologic mechanisms associated with thrombus formation in the absence of a ruptured plaque including plaque erosion, small (and undetectable) plaque ruptures, and potentially un-ruptured TCFAs with superimposed thrombosis.
Dynamic Nature of Atherosclerotic Lesions
The ability of imaging modalities to serially image lesions has better defined the natural history of putative high risk lesions, those predictive features of future clinical events, as well as the response of vulnerable features to medical therapy. Two serial VH-IVUS studies added important insights into dynamic, plaque compositional changes over time. In an evaluation of 99 patients (77 % with stable angina), followed for 12 months, 12 TCFAs developed, six from pathological intimal thickening and six from thick-cap fibroatheromas, while baseline fibrous and fibrocalcific plaque progressed less often. Of the 20 baseline TCFA lesions, 15 (75 %) regressed on follow up, with 13 becoming thick fibrous cap atheromas and two becoming fibrotic lesions [44••]. On the other hand, a serial study of patients presenting with a STEMI showed that only 22 % of non-culprit TCFAs regressed after 13 months, while the remainder demonstrated a significant progression in lesion severity and an increase in necrotic core size [45••]. The varying results of the two studies implicate a role of inflammation in STEMI patients leading to a persistent presence of high-risk lesions.
In a prospective study of 552 coronary artery disease patients who underwent angioscopy and were followed for 57.3 ± 22.1 months, the presence of multiple yellow plaques per vessel was an independent risk factor for subsequent acute coronary events [46]. Of 30 patients with non-culprit ruptured plaques, 23 % were healed at ≤12 months and 55 % were healed at >12 months. Patients with higher CRP levels had lower rates of plaque healing, illustrating the role of inflammation and dynamic plaque changes. Healed ruptured plaques showed progressive angiographic stenosis on follow up, verifying findings of VH-IVUS studies showing multiple layers of healed plaque rupture within advanced angiographic severe stenoses [47].
Although there is a lack of serial NIRS studies in humans, in a study of diabetic/hypercholesterolemic pigs that develop vulnerable, human-like coronary lesions, NIRS lesions showing high lipid core plaque at 6 months predicted the development of plaques with vulnerable features at 9 months including high-risk fibroatheroma with larger plaque area, larger necrotic core area, and thinner fibrous caps with a high concentration of inflammatory cells [48]. Clinical studies evaluating the ability of NIRS to predict high risk lesions in humans are currently underway.
Medical therapy may impact vulnerable plaque features associated with clinical events. In a natural history study of 32 patients with ACS treated with contemporary pharmacologic therapy who underwent CTA at baseline and at a median of 39 months later, the minimum lumen diameter increased by 0.15 mm (−0.09 to 0.24, p = 0.039) while total atheroma volume increased by 6.7 % (p = 0.029) and was accompanied by positive remodeling in 53 % of patients. Patients with subsequent coronary events had a higher normalized total atheroma volume at baseline (969.72 mm3 vs. 810.77 mm3, p = 0.010) [49]. Statin use has been shown to decrease total plaque volume and low attenuation plaque volume by CTA [50]. Multiple IVUS studies evaluating the effect of statins show a favorable change in vulnerable plaque features including healing of plaque rupture and an increase in lumen area [51], a decrease in atheroma volume [52], and a decrease in fibro-fatty volume with an increase in fibrous tissue volume [53]. Statin treatment was also shown, by OCT, to increase fibrous cap thickness, lending imaging evidence for the plaque stabilization effects of statins as a mechanism for decreasing acute coronary events [54].
Predicting Future Ischemic Events
Prospective imaging studies have identified those high-risk plaque features which correlate with subsequent clinical events or plaque progression, thereby establishing a paradigm for the prophylactic treatment of lesions before they cause clinical events. For example, plaques demonstrating positive remodeling and low-attenuation by CTA have a higher risk of causing ACS. Motoyama et. al followed 1,059 patients for 27 ± 10 months and showed that 22.2 % of patients (45/1,059) with remodeled, low-attenuation plaques developed ACS compared to 3.7 % of patients (1/27) possessing plaques which displayed only one of the features. Of the 820 patients with neither feature, the rate for developing an ACS event was 0.5 %. Positive remodeling and/or low-attenuation were independent predictors of ACS events (HR: 22.8, 95 % CI: 6.9 to 75.2, p < 0.001) [55]. Yamamoto et. al followed 511 patients for 3.3 ± 1.2 years. The presence of non-calcified atherosclerotic plaques by CTA with both low attenuation and positive remodeling predicted cardiovascular events (HR:11.2, 95 % CI, 3.71 – 36.7, p < 0.0001) [56].
The largest invasive natural history study on vulnerable plaque was PROSPECT which followed 697 patients presenting with an ACS. After PCI of the culprit lesion, three-vessel VH-IVUS was performed. Follow-up angiography was not mandated. At 3 years the cumulative major adverse cardiovascular event rate was 20.4 % of which 11.6 % were in non-culprit lesions. Most non-culprit lesions that caused subsequent events were angiographically mild at baseline (mean diameter stenosis of 32 %), but were characterized by a plaque burden >70 % by IVUS, a minimal luminal area of 4.0 mm2 or less, or were classified as a VH-TCFA [57••]. When the lesion possessed all three features, the subsequent event rate was 18.2 %. In an analysis of all 3,115 non-culprit lesions at baseline, increasing severity of angiographic stenosis correlated with increased prevalence of high-risk features including IVUS minimum luminal areas of ≤4 mm2, IVUS plaque burden over 70 %, greater necrotic core volume, and VH-TCFA [58]. The VIVA study, a similar but smaller prospective study, corroborated these findings. Patients with stable or unstable coronary disease (n = 170) underwent three-vessel VH-IVUS and were followed for a median of 1.7 years. The presence of a VH-TCFA (HR: 7.53, p = 0.038) and a plaque burden >70 % (HR: 8.13, p = 0.011) predicted future cardiac events [59].
The PREDICTION study used VH-IVUS plaque features in addition to local hemodynamic characteristics to predict future non-culprit lesion coronary events in 506 patients with an ACS event treated with PCI. Baseline three vessel VH-IVUS study and assessment of endothelial shear stress using a computational model was performed. A subset of 374 patients had a follow up study at 6 – 10 months. An increase in plaque area and a decrease in lumen area of non-culprit lesions were predicted by a large baseline plaque burden while low endothelial shear stress predicted a decrease in lumen area. Adding low endothelial shear stress to large baseline plaque burden increased the positive predictive value to predict progression of an obstruction leading to angina from 22 % to 41 %. If both features were absent, the negative predictive value of plaque progression was 92 % [60••].
Conclusion
New imaging modalities have advanced the field of vulnerable atherosclerotic plaque research. Many findings determined by histological evaluation of post mortem samples have been verified in vivo and further advanced by invasive and non-invasive imaging. The main features that characterize a vulnerable plaque and predict future coronary events are greater plaque burden, larger lipid and necrotic core, thinner fibrous caps, and inflammatory cells infiltrating the fibrous cap. However, a significant percentage of lesions causing events are not the classically defined TCFA. Arteries with a ruptured plaque have been shown to possess a number of high-risk lesions supporting the hypothesis of pan-coronary inflammation. In vivo imaging has shown the dynamic nature of vulnerable plaque, with both plaque progression and regression being common findings. With the further development of non-invasive imaging that minimizes radiation and cost, and invasive imaging systems that minimize risk, it is conceivable that a screening strategy of asymptomatic patients may identify a high-risk population that would benefit from aggressive prophylactic medical or interventional treatment.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18–e209.
Davies MJ. The pathophysiology of acute coronary syndromes. Heart. 2000;83(3):361–6.
Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20(5):1262–75.
Narula J, Nakano M, Virmani R, et al. Histopathologic characteristics of atherosclerotic coronary disease and implications of the findings for the invasive and noninvasive detection of vulnerable plaques. J Am Coll Cardiol. 2013;61(10):1041–51. Further refinement of the definition of a thin-cap fibroatheroma by necropsy evaluation..
Ohayon J, Finet G, Gharib AM, et al. Necrotic core thickness and positive arterial remodeling index: emergent biomechanical factors for evaluating the risk of plaque rupture. Am J Physiol Heart Circ Physiol. 2008;295(2):H717–727.
Arbustini E, Grasso M, Diegoli M, et al. Coronary atherosclerotic plaques with and without thrombus in ischemic heart syndromes: a morphologic, immunohistochemical, and biochemical study. Am J Cardiol. 1991;68(7):36B–50B.
Voros S, Rinehart S, Qian Z, et al. Coronary atherosclerosis imaging by coronary CT angiography: current status, correlation with intravascular interrogation and meta-analysis. JACC Cardiovasc Imaging. 2011;4(5):537–48.
Rogers IS, Tawakol A. Imaging of coronary inflammation with FDG-PET: feasibility and clinical hurdles. Curr Cardiol Rep. 2011;13(2):138–44.
Rosa GM, Bauckneht M, Masoero G, et al. The vulnerable coronary plaque: update on imaging technologies. Thromb Haemost. 2013;110(4):706–22.
Prati F, Regar E, Mintz GS, et al. Expert review document on methodology, terminology, and clinical applications of optical coherence tomography: physical principles, methodology of image acquisition, and clinical application for assessment of coronary arteries and atherosclerosis. Eur heart J. 2010;31(4):401–15.
Yabushita H, Bouma BE, Houser SL, et al. Characterization of human atherosclerosis by optical coherence tomography. Circulation. 2002;106(13):1640–5.
Tearney GJ, Yabushita H, Houser SL, et al. Quantification of macrophage content in atherosclerotic plaques by optical coherence tomography. Circulation. 2003;107(1):113–9.
Sanon S, Dao T, Sanon VP, Chilton R. Imaging of vulnerable plaques using near-infrared spectroscopy for risk stratification of atherosclerosis. Curr Atheroscler Rep. 2013;15(2):304.
Thieme T, Wernecke KD, Meyer R, et al. Angioscopic evaluation of atherosclerotic plaques: validation by histomorphologic analysis and association with stable and unstable coronary syndromes. J Am Coll Cardiol. 1996;28(1):1–6.
Takano M, Jang IK, Inami S, et al. In vivo comparison of optical coherence tomography and angioscopy for the evaluation of coronary plaque characteristics. Am J Cardiol. 2008;101(4):471–6.
Ambrose JA, Tannenbaum MA, Alexopoulos D, et al. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol. 1988;12(1):56–62.
Cutlip DE, Chhabra AG, Baim DS, et al. Beyond restenosis: five-year clinical outcomes from second-generation coronary stent trials. Circulation. 2004;110(10):1226–30.
Glaser R, Selzer F, Faxon DP, et al. Clinical progression of incidental, asymptomatic lesions discovered during culprit vessel coronary intervention. Circulation. 2005;111(2):143–9.
Hoffmann U, Moselewski F, Nieman K, et al. Noninvasive assessment of plaque morphology and composition in culprit and stable lesions in acute coronary syndrome and stable lesions in stable angina by multidetector computed tomography. J Am Coll Cardiol. 2006;47(8):1655–62.
Pflederer T, Marwan M, Schepis T, et al. Characterization of culprit lesions in acute coronary syndromes using coronary dual-source CT angiography. Atherosclerosis. 2010;211(2):437–44.
Hernando L, Corros C, Gonzalo N, et al. Morphological characteristics of culprit coronary lesions according to clinical presentation: insights from a multimodality imaging approach. Int J Cardiovasc Imaging. 2013;29(1):13–21.
Fujii K, Kobayashi Y, Mintz GS, et al. Intravascular ultrasound assessment of ulcerated ruptured plaques: a comparison of culprit and nonculprit lesions of patients with acute coronary syndromes and lesions in patients without acute coronary syndromes. Circulation. 2003;108(20):2473–8.
Madder RD, Smith JL, Dixon SR, Goldstein JA. Composition of target lesions by near-infrared spectroscopy in patients with acute coronary syndrome versus stable angina. Circ Cardiovasc Interv. 2012;5(1):55–61. One of the first clinical papers to use NIRS showing that patients with acute coronary syndromes have lesions with high lipid core plaque compared to patients with stable angina..
Madder RD, Goldstein JA, Madden SP, et al. Detection by Near-Infrared Spectroscopy of Large Lipid Core Plaques at Culprit Sites in Patients With Acute ST-Segment Elevation Myocardial Infarction. JACC Cardiovasc Interv. 2013;6(8):838–46.
Jang IK, Tearney GJ, MacNeill B, et al. In vivo characterization of coronary atherosclerotic plaque by use of optical coherence tomography. Circulation. 2005;111(12):1551–5.
Kubo T, Imanishi T, Kashiwagi M, et al. Multiple coronary lesion instability in patients with acute myocardial infarction as determined by optical coherence tomography. Am J Cardiol. 2010;105(3):318–22.
Yonetsu T, Kakuta T, Lee T, et al. In vivo critical fibrous cap thickness for rupture-prone coronary plaques assessed by optical coherence tomography. Eur Heart J. 2011;32(10):1251–9.
Tanaka A, Imanishi T, Kitabata H, et al. Morphology of exertion-triggered plaque rupture in patients with acute coronary syndrome: an optical coherence tomography study. Circulation. 2008;118(23):2368–73.
Toutouzas K, Tsiamis E, Karanasos A, et al. Morphological characteristics of culprit atheromatic plaque are associated with coronary flow after thrombolytic therapy: new implications of optical coherence tomography from a multicenter study. JACC Cardiovasc Interv. 2010;3(5):507–14. Study showing that an acute myocardial infarction can be caused by rupture of fibroatheromas not specifically defined as TCFAs..
Ino Y, Kubo T, Tanaka A, et al. Difference of culprit lesion morphologies between ST-segment elevation myocardial infarction and non-ST-segment elevation acute coronary syndrome: an optical coherence tomography study. JACC Cardiovasc Interv. 2011;4(1):76–82.
Kato K, Yonetsu T, Kim SJ, et al. Nonculprit plaques in patients with acute coronary syndromes have more vulnerable features compared with those with non-acute coronary syndromes: a 3-vessel optical coherence tomography study. Circ Cardiovasc Imaging. 2012;5(4):433–40.
Hong MK, Mintz GS, Lee CW, et al. Comparison of virtual histology to intravascular ultrasound of culprit coronary lesions in acute coronary syndrome and target coronary lesions in stable angina pectoris. Am J Cardiol. 2007;100(6):953–9.
Fujii K, Carlier SG, Mintz GS, et al. Intravascular ultrasound study of patterns of calcium in ruptured coronary plaques. Am J Cardiol. 2005;96(3):352–7.
Kashiwagi M, Tanaka A, Kitabata H, et al. Feasibility of noninvasive assessment of thin-cap fibroatheroma by multidetector computed tomography. JACC Cardiovasc Imaging. 2009;2(12):1412–9.
Kroner ES, van Velzen JE, Boogers MJ, et al. Positive remodeling on coronary computed tomography as a marker for plaque vulnerability on virtual histology intravascular ultrasound. Am J Cardiol. 2011;107(12):1725–9.
Maurovich-Horvat P, Schlett CL, Alkadhi H, et al. The napkin-ring sign indicates advanced atherosclerotic lesions in coronary CT angiography. JACC Cardiovasc Imaging. 2012;5(12):1243–52. While coronary computed tomography does not have the resolution of invasive imaging modalities to discern fibrous cap thickness, necrotic core size, and other classic histologic features of high risk lesions, there are other unique imaging features that are associated with high risk lesions. In this paper, a ring-like pattern of low attenuation termed the napkin-ring sign was shown to identify high risk lesions with a high specificity compared to histological evaulation..
Nishio M, Ueda Y, Matsuo K, et al. Detection of disrupted plaques by coronary CT: comparison with angioscopy. Heart. 2011;97(17):1397–402.
Kitabata H, Tanaka A, Kubo T, et al. Relation of microchannel structure identified by optical coherence tomography to plaque vulnerability in patients with coronary artery disease. Am J Cardiol. 2010;105(12):1673–8.
Uemura S, Ishigami K, Soeda T, et al. Thin-cap fibroatheroma and microchannel findings in optical coherence tomography correlate with subsequent progression of coronary atheromatous plaques. Eur Heart J. 2012;33(1):78–85.
MacNeill BD, Jang IK, Bouma BE, et al. Focal and multi-focal plaque macrophage distributions in patients with acute and stable presentations of coronary artery disease. J Am Coll Cardiol. 2004;44(5):972–9.
Rogers IS, Nasir K, Figueroa AL, et al. Feasibility of FDG imaging of the coronary arteries: comparison between acute coronary syndrome and stable angina. JACC Cardiovasc Imaging. 2010;3(4):388–97.
Kim SW, Hong YJ, Mintz GS, et al. Relation of ruptured plaque culprit lesion phenotype and outcomes in patients with ST elevation acute myocardial infarction. Am J Cardiol. 2012;109(6):794–9. This paper showed that lower risk lesions (non-TCFAs) are associated with plaque rupture, including thick cap fibroatheroma, fibro-calcified plaque, and pathologic intimal thickening..
Sanidas EA, Maehara A, Mintz GS, et al. Angioscopic and virtual histology intravascular ultrasound characteristics of culprit lesion morphology underlying coronary artery thrombosis. Am J Cardiol. 2011;107(9):1285–90.
Kubo T, Maehara A, Mintz GS, et al. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol. 2010;55(15):1590–7. This paper shows that in patients with stable angina, high risk plaques over time can regress to lower risk features, with 75 % of thin cap fibroatheromas showing signs of stabilization or healing over time..
Zhao Z, Witzenbichler B, Mintz GS, et al. Dynamic nature of nonculprit coronary artery lesion morphology in STEMI: a serial IVUS analysis from the HORIZONS-AMI trial. JACC Cardiovasc Imaging. 2013;6(1):86–95. As opposed to the Kubo (2010) et al. paper showing that patients with stable angina have high risk plaques that tend to regress over time, in a more high risk patient population presenting with ST elevation myocardial infarctions, lesions tend advance to higher risk features over time. These two papers would suggest that patient specific features, such as a potential background milieu of higher inflammation, will determine whether high risk lesions will progress and cause clinical syndromes or regress over time..
Ohtani T, Ueda Y, Mizote I, et al. Number of yellow plaques detected in a coronary artery is associated with future risk of acute coronary syndrome: detection of vulnerable patients by angioscopy. J Am Coll Cardiol. 2006;47(11):2194–200.
Takano M, Inami S, Ishibashi F, et al. Angioscopic follow-up study of coronary ruptured plaques in nonculprit lesions. J Am Coll Cardiol. 2005;45(5):652–8.
Patel D, Hamamdzic D, Llano R, et al. Subsequent development of fibroatheromas with inflamed fibrous caps can be predicted by intracoronary near infrared spectroscopy. Arterioscler Thromb Vasc Biol. 2013;33(2):347–53.
Papadopoulou SL, Neefjes LA, Garcia-Garcia HM, et al. Natural history of coronary atherosclerosis by multislice computed tomography. JACC Cardiovasc Imaging. 2012;5(3 Suppl):S28–37.
Inoue K, Motoyama S, Sarai M, et al. Serial coronary CT angiography-verified changes in plaque characteristics as an end point: evaluation of effect of statin intervention. JACC Cardiovasc Imaging. 2010;3(7):691–8.
Hong MK, Mintz GS, Lee CW, et al. Serial intravascular ultrasound evidence of both plaque stabilization and lesion progression in patients with ruptured coronary plaques: effects of statin therapy on ruptured coronary plaque. Atherosclerosis. 2007;191(1):107–14.
Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295(13):1556–65.
Nasu K, Tsuchikane E, Katoh O, et al. Effect of fluvastatin on progression of coronary atherosclerotic plaque evaluated by virtual histology intravascular ultrasound. JACC Cardiovasc Interv. 2009;2(7):689–96.
Takarada S, Imanishi T, Kubo T, et al. Effect of statin therapy on coronary fibrous-cap thickness in patients with acute coronary syndrome: assessment by optical coherence tomography study. Atherosclerosis. 2009;202(2):491–7.
Motoyama S, Sarai M, Harigaya H, et al. Computed tomographic angiography characteristics of atherosclerotic plaques subsequently resulting in acute coronary syndrome. J Am Coll Cardiol. 2009;54(1):49–57.
Yamamoto H, Kitagawa T, Ohashi N, et al. Noncalcified atherosclerotic lesions with vulnerable characteristics detected by coronary CT angiography and future coronary events. J Cardiovasc Comput Tomogr. 2013;7(3):192–9.
Stone GW, Maehara A, Lansky AJ, et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364(3):226–35. A pivotal natural history study using VH-IVUS and following 697 patients who presented with an acute coronary syndrome for three years, this study defined high risk plaque features assocated with subsequent events caused by initially non-culprit lesions. Most of these lesions were angiographically mild at baseline (mean diameter stenosis of 32 %) but had a plaque burden over 70 %, a minimal luminal area of 4.0 mm^2, or were classified as a thin cap fibroatheroma. If a lesion had all three high risk features, the future event rate rose to 18.2 %..
Yun KH, Mintz GS, Farhat N, et al. Relation between angiographic lesion severity, vulnerable plaque morphology and future adverse cardiac events (from the Providing Regional Observations to Study Predictors of Events in the Coronary Tree study). Am J Cardiol. 2012;110(4):471–7.
Calvert PA, Obaid DR, O'Sullivan M, et al. Association between IVUS findings and adverse outcomes in patients with coronary artery disease: the VIVA (VH-IVUS in Vulnerable Atherosclerosis) Study. JACC Cardiovasc Imaging. 2011;4(8):894–901.
Stone PH, Saito S, Takahashi S, et al. Prediction of progression of coronary artery disease and clinical outcomes using vascular profiling of endothelial shear stress and arterial plaque characteristics: the PREDICTION Study. Circulation. 2012;126(2):172–81. In this study of 506 patients presenting with an acute coronary syndrome, three vessel VH-IVUS as well as a computational assessment of enothelial shear stress was performed and a subset of 374 patients had a routine follow up study in 6–10 months. Similar to PROSPECT, a large baseline plaque burden predicted the progression of lesions. Interestingly, adding the low endothelial shear stress computation to baseline plaque burden doubled the positive predictive value to predict progression from 22 % to 41 %..
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Robert S. Fenning and Robert L. Wilensky declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Coronary Heart Disease
Rights and permissions
About this article
Cite this article
Fenning, R.S., Wilensky, R.L. New Insights into the Vulnerable Plaque from Imaging Studies. Curr Atheroscler Rep 16, 397 (2014). https://doi.org/10.1007/s11883-014-0397-1
Published:
DOI: https://doi.org/10.1007/s11883-014-0397-1