
Abstract
The use of gene therapy in the treatment of primary immune deficiencies (PID) has advanced significantly in the last decade. Clinical trials for X-linked severe combined immunodeficiency, adenosine deaminase deficiency (ADA), chronic granulomatous disease, and Wiskott-Aldrich syndrome have demonstrated that gene transfer into hematopoietic stem cells and autologous transplant can result in clinical improvement and is curative for many patients. Unfortunately, early clinical trials were complicated by vector-related insertional mutagenic events for several diseases with the exception of ADA-deficiency SCID. These results prompted the current wave of clinical trials for primary immunodeficiency using alternative retro- or lenti-viral vector constructs that are self-inactivating, and they have shown clinical efficacy without leukemic events thus far. The field of gene therapy continues to progress, with improvements in viral vector profiles, stem cell culturing techniques, and site-specific genome editing platforms. The future of gene therapy is promising, and we are quickly moving towards a time when it will be a standard cellular therapy for many forms of PID.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary immunodeficiency diseases (PID) are inherited disorders of the immune system resulting in predisposition to infection often accompanied by immune dysregulation or autoimmunity. They can be broadly classified as innate immunodeficiencies, antibody deficiencies, combined B and T lymphocyte immunodeficiencies with or without syndromic features, defects of phagocytes, complement deficiencies, and diseases of immune dysregulation or autoinflammation. The list of gene defects responsible for these conditions is rapidly expanding as improved diagnostic modalities have led to a more extensive understanding and characterization of the immune system [1]. For those with life-threatening PID such as severe combined immunodeficiency (SCID), allogeneic hematopoietic stem cell transplant (HSCT) has been the main curative therapy. Although advancements in supportive care such as prevention of infections, HLA typing, and conditioning regimens have significantly improved HSCT outcomes in recent years, especially for those who are transplanted when young and relatively healthy with well-matched donors, HSCT can be complicated in those who lack matched donors or have preexisting infection [2••, 3]. For these patients, optimal therapy may be achieved with autologous HSCT using gene modified stem cells.
The concept of gene therapy was born in the 1980s with the ability to manipulate genetic material in the test tube and grew further with the development of viral vectors to stably integrate modified genes into cells. In 1990, the first clinical trial of gene therapy was attempted in a 4-year-old girl with SCID due to deficiency of the purine catabolic enzyme adenosine deaminase (ADA). Although infusions of patient T cells treated with a gamma-retroviral vector containing normal ADA complementary DNA (cDNA) were safe and resulted in production of endogenous ADA enzyme, these effects required multiple rounds of T cell infusions and were not sufficient to remove the requirement for exogenous ADA enzyme replacement [4]. Nevertheless, it established a foundation that recombinant DNA could be used to treat disease safely and, potentially, permanently. Since this time, gene therapy has become a convincing therapy for PID with significant advancements in the understanding of viral vectors, manipulation of HSCs, and implementation of conditioning regimens (Table 1).
Adenosine Deaminase Deficiency SCID
The first immunodeficiency for which a specific molecular defect was described is adenosine deaminase deficiency, an autosomal recessive disorder manifesting as T, B, and NK cell dysfunction. It accounts for about 15 % of all SCID cases at an overall incidence of 1 in 500,000 live births. The ADA enzyme is ubiquitously found intracellularly and on the surface of all cells and is crucial for the metabolism of adenosine and 2-deoxyadenosine. Enzyme activity is highest in lymphoid tissues, particularly the thymus, and the lack of ADA results in the accumulation of toxic purine metabolites particularly at sites of increased cell turnover, such as the bone marrow and thymus where lymphocytes undergo apoptotic death during differentiation and selection. Enzyme replacement therapy with polyethylene glycol-modified bovine ADA provides lifesaving metabolic detoxification and rapid immune reconstitution, but it is non-curative and patients may remain susceptible to infections due to suboptimal B and T cell recovery [5]. Over time, the response to enzyme replacement therapy becomes progressively blunted, manifesting particularly as a decreasing B cell compartment [6].
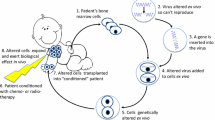
HSCT from an unaffected, matched sibling donor is the treatment of choice for a definitive cure. Success rates are significantly reduced in patients who receive matched unrelated and haploidentical donors or for those who have an infection at the time of transplant [2••, 7]. For those without well-matched donors, gene therapy with autologous stem cells is becoming a standard of care. (Fig. 1) The first gene therapy trials targeted delivery of the corrective ADA gene to either T lymphocytes or cord blood/bone marrow progenitor cells using murine gamma-retroviral vectors while patients were maintained on ERT without the use of cytoreductive conditioning [4, 8–11]. Despite low but detectable gene-marked cells after therapy, ADA expression was not high enough to confer significant clinical benefit. Nevertheless, it established safety and the potential for gene therapy as a treatment modality in ADA SCID. Since then, there has been further development in gene therapy clinical trials for this disease.

Schema of ex vivo autologous gene therapy
The next wave of clinical trials used Moloney murine leukemia virus-derived replication-deficient recombinant retroviruses to deliver normal ADA cDNA. In addition to optimized gene transduction methods, nonmyeloablative doses of busulfan or melphalan were used to create space in the bone marrow. In addition, ERT was discontinued either shortly before or right at the time of reinfusion of modified stem cells. This change was based on a report of two patients for whom PEG-ADA was not available suggesting that the toxic metabolic environment in the face of PEG-ADA discontinuation provided a selective pressure for gene-modified progeny. Of ten patients treated in Italy, 9/10 patients maintained lasting immune reconstitution, with eight of ten and five of ten off ERT and IVIG, respectively [12••]. In the UK, six patients were treated in a similar fashion with four of six off ERT and three of six off IVIG [13]. In the USA, ten ADA SCID patients were treated in a collaborative study between The National Human Genome Institute, NIH and Children’s Hospital Los Angeles, and later the UCLA Mattel Children’s Hospital. These patients received a combination of two similar retroviral vectors; four of these patients remained on ERT and received no bone marrow conditioning while six underwent busulfan preconditioning without ERT. [14] Only those who received marrow conditioning demonstrated clinically relevant levels of gene-corrected T cells with adequate ADA enzyme activity and immune recovery, emphasizing the importance of nonmyeloablative pre-transplantation conditioning in gene therapy for ADA SCID. Subsequently, ten additional patients were treated in a phase II study using autologous CD34+ cells modified with a single ADA gamma-retroviral vector driven by the MND promotor after conditioning with busulfan and ERT cessation. Nine of ten patients remain off ERT and three have discontinued IVIG. The one patient who remains on ERT was the oldest patient, treated at 15 years [15].
To date, from the above listed trials, there have been at least 42 patients treated with gamma-retroviral ADA gene therapy throughout the USA and Europe with 100 % survival. In contrast to all other gene therapy trials for primary immune deficiency including X-SCID, X-CGD, and WAS, none of the ADA SCID patients have developed abnormal clonal expansions or leukoproliferative disorders despite the use of very similar vectors. Genome-wide analyses of retroviral vector integrations in five patients treated with ADA SCID gene therapy demonstrated similar insertion site profiles to other trials near proto-oncogenes (including LMO2), yet there was no skewing of the T cell repertoire, nor was there an associated clonal selection or expansion in vivo [16]. Although the reasons for these differences are not clear, there is currently no evidence to show that the transgene itself is responsible for myeloproliferative complications [17]. It has been postulated that ADA enzyme production may cross-rescue ADA-deficient lymphocytes, thereby placing less replicative stress on gene-corrected cells. In addition, myeloid cell abnormalities in patients with ADA SCID may protect them from malignant transformation [18].
Although there has been no insertional genotoxicity observed in ADA patients treated with gamma-retroviral vectors, continued concerns regarding leukemogenesis have led to the current generation of trials using self-inactivating HIV-1-based lentiviral vectors. HIV tat and accessory proteins (vpu, vpr, nef, vif) have been deleted from the vector packaging plasmids to create a safer vector profile, and removal of the U3 region enhancers from the long terminal repeats (LTR) is accomplished by the self-inactivation (SIN) mechanism. Lentiviral vectors are usually pseudotyped with the vesicular stomatitis virus glycoprotein which uses the broadly expressed LDL receptor for cell entry and results in diverse cell tropism. Importantly, lentiviral vectors can transduce non-dividing cells, such as quiescent hematopoietic stem cells. In addition, ongoing work in murine models challenged the idea that withdrawal of ERT provided a survival advantage for transduced cells [19]. Given no differences in gene-marked lymphocytes amongst mice with or without ERT, the current trials in the US and UK leave subjects on ERT until 30 days post transplant. At the time of this publication, there have been at least 30 ADA SCID patients treated with lentiviral vectors in the USA and the UK. Excellent immune reconstitution is being achieved in the treated patients with no vector-related complications to date, with up to 3 years follow-up [13].
Gene Therapy Trials for X-Linked Severe Combined Immunodeficiency (X-SCID)
Gene therapy trials for X-SCID followed discovery of the responsible gene in 1993 [20]. Defects in the interleukin-2 receptor subunit gamma (IL2RG) gene on the X-chromosome which encodes the cytokine receptor common γ chain account for the most common form of SCID. The common γ chain is an essential component of receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 and results in a lack of both T cells and natural killer cells with present but immature B cells and no humoral immunity. The first clinical trial of X-SCID opened in 1999 in France (nine patients and one treated in Australia) and was closely followed in 2001 by a similar trial in the UK (four patients). Both sites enrolled typical X-SCID patients who lacked HLA-identical bone marrow donors and treated patients with autologous bone marrow CD34+ HSCs transduced ex vivo with a murine gamma-retroviral vector in which the γc gene was under control of the viral LTR. Gene-modified cells were returned to patients without undergoing cytoreductive conditioning. Treatment with γc gene therapy resulted in stable reconstitution of both cellular and humoral immunity, and patients were able to combat typical childhood infections such as varicella zoster and resume normal growth and development [21••, 22].
Although there was no apparent decrease in T cell number or repertoire diversity over time, four patients in the French trial and one in the UK trial developed T cell acute lymphoblastic leukemia 31–68 months after gene therapy [23, 24•]. In four of these patients, the gammaretroviral vector in the leukemic clone was integrated near the LIM domain-only 2 (LMO2) proto-oncogene and activated its transcription. In the fifth patient, the vector integrated near CCND2, which encodes cyclin D2 and is a known oncogene in lymphoid cells [24•]. In addition, chromosomal translocation events were detected in blast cells, indicating that multiple events had led to leukemogenesis. Sustained remission with normal immunity was achieved with chemotherapy in four of the five patients, while the fifth patient ultimately died after chemotherapy and bone marrow transplant. Although the initial promising results from these trials were overshadowed by serious adverse complications, the overall outcome of both studies was very positive with 19 of 20 patients alive at last follow-up, 18 with immune function due solely to gene therapy.
Subsequently, modifications were made to the original vector backbone which contained duplicated viral enhancer sequences within the LTRs. A self-inactivating gammaretroviral vector-expressing IL2RG cDNA from a promoter lacking enhancers to trans-activate cellular genes, the human elongation factor 1α (EF1α) short promoter, has been used in nine boys in parallel European and American trials without preparative conditioning [25••]. At follow-up, eight of nine children are living, while one died from a preexisting adenoviral infection prior to immune reconstitution. Of those surviving, seven had stable engraftment with functional T cells and resolution of infections. Importantly, integration analysis demonstrated no clonal skewing and less clustering of insertion sites near lymphoid proto-oncogenes. This trial remains open and patient recruitment is ongoing. An additional two-site clinical trial using a self-inactivating lentiviral vector for XSCID has also opened in the USA. Thus far, two subjects, both 24 years of age at time of treatment, are alive with clinical improvement at 1 year and 4 months follow-up [26].
Gene Therapy for X-Linked Chronic Granulomatous Disease (X-CGD)
Following the positive results of trials for X-linked and ADA SCID, clinical trials were developed for X-CGD to correct defects in the CYBB gene, which encodes gp91phox, an essential component of NADPH-oxidase in phagocytic cells. Absence or malfunction of any of the NADPH-oxidase subunits results in a primary immunodeficiency characterized by recurrent, life-threatening bacterial and fungal infections of the liver, lung, bones, and skin with severe inflammatory complications. The most common form of CGD is X-linked, accounting for about 60 % of all cases. Available therapies include life-long antibiotic prophylaxis, immunomodulation with interferon-γ, and allogeneic HSCT. Although allogeneic stem cell transplant for CGD has improved considerably, severe side-effects such as graft-versus-host disease and inflammatory exacerbations are often seen in those with pre-existing, ongoing infections indicating the need for alternative therapies [27•].
The first clinical trials with gene modified cells for CGD at the National Institutes of Health and Indiana University used gamma retroviral vectors to deliver normal human gp91phox. Although there were no severe adverse events and gene-marked cells were detectable at low levels for months after treatment, resulting superoxide levels were not sufficient to produce significant clinical improvement. Notably, a common denominator amongst these early trials was the lack of bone marrow conditioning. Following these results, three patients were treated at the NIH using reduced intensity conditioning prior to infusion of gp91phox vector-treated HSCs. Although functionally corrected cells dropped from 24 to 1.1 % at 34 months post therapy, there was still resolution of infections at this low level of transgene expression, indicating that even a small amount of normal superoxide production could result in meaningful clinical outcomes. Levels were 0.03 % in a second patient who had partial control of infections at 11 months follow-up and undetectable in the third who died of invasive fungal infection 6 months post treatment [28•].
Worldwide, nine additional X-CGD patients were treated in Frankfurt, Zurich, London, and Seoul with similar protocols using gamma-retroviral vectors and reduced intensity conditioning. There was initial clinical benefit in all of these patients, although of those treated, three developed genotoxicity due to insertional activation of growth promoting genes, particularly PRDM16 and MDS1/EVI1. Overexpression of EVI1 led to the development of myelodysplasia with monosomy seven in two patients. In addition, there was a gradual loss of functional gene-marked cells at around 8 months after gene therapy due to epigenetic inactivation of the retroviral promoter. In patients where there were no leukemogenic events, there was also no significant engraftment after 3 months post gene therapy.
Many important lessons were learned from these early XCGD gene therapy trials. First, they established that gene therapy holds the potential to bring severe, non-responsive infections under control. Even with low rates of gene marking after treatment, patients experienced clinical benefits due to superoxide productions from <1 % of cells. Secondly, bone marrow conditioning is a particularly important issue for gene therapy in XCGD patients. Previous publications of allogeneic HSCT have shown that nearly ablative busulfan doses were safe in patients with XCGD, and high level myelosuppression would likely be necessary to ensure long-lasting correction [27•]. Lastly, leukemic events in three treated patients emphasized the importance of choosing a vector that could transduce primitive HSC with high efficiency while maintaining a safe profile of vector integration.
There are currently two parallel gene therapy trials in Europe and the USA based on these rationales with safer lentiviral vectors. There has been one child with significant disease burden (invasive liver, brain, abdominal, and pulmonary infections, and inflammatory complications) treated in Europe. He was stable until around 3 months post gene therapy, but then died from respiratory complications due to pre-existing conditions. The US trial is currently open and is anticipating treatment of the first patient by the end of 2015.
Gene Therapy for Wiskott-Aldrich Syndrome
Wiskott-Aldrich Syndrome (WAS) is an X-linked disorder classically described as a triad of increased susceptibility to infections, microthrombocytopenia, and eczema due to defects in the WAS gene that encodes a cytoplasmic protein affecting actin polymerization in hematopoietic cells. Functional WAS protein is required for leukocyte function, migration, and formation of the immunologic synapse. In addition, autoimmunity is often a complicating factor due to inadequate T regulatory cell function, autoreactive B cells, and defective clearance of apoptotic cells [29]. Despite improvements in clinical care, outcomes for patients with classic WAS remain poor with a median life expectancy of 15 years for those who do not receive curative bone marrow transplant. Irrespective of donor type, overall survival from allogeneic HSCT is around 84 % but higher when there is a HLA-identical sibling donor. Autoimmune complications are more frequent in those who do not achieve complete chimerism post allogeneic transplant [30]. Given these factors, treatment with gene-corrected autologous HSCs was explored as a beneficial alternative therapy.
Based on extensive preclinical studies, the first clinical trial for WAS was conducted between 2006 and 2009 in Germany and included ten patients with severe WAS. Patients received reduced intensity doses of busulfan and autologous CD34+ cells transduced with a WASP-expressing gammaretroviral vector [31]. After gene therapy, there was a large increase in the proportion of corrected lymphoid cells due to the proliferative advantage of cells that express functional WAS protein. Reconstitution of lymphocyte function was demonstrated through T cell proliferative responses and antibody titers after vaccination clinically translating to reduced frequency and severity of infections. In addition, all but one patient had significant and sustained increases in platelet counts and normalization of platelet size providing resolution of hemorrhagic diatheses within 1 year of treatment [31, 32]. However, vector insertion site analysis revealed marked and similar clustering with 17 of the 25 most affected genomic regions previously identified as proto-oncogenes. Four of these included MDS1-EVI1, PRDM16, LMO2, and CCND2, all of which were reported as common integration sites in clonal expansions in the X-SCID and X-CGD gene therapy trials. Unfortunately, between 14 months and 5 years after gene therapy, seven of ten patients developed hematologic malignancies, six of which were LMO2 related. Affected patients were treated with chemotherapy and allogeneic HSCT, but two patients died from leukemia.
Subsequently, there have been three gene therapy trials in France, England, Italy, and the USA using a self-inactivating lentiviral vector and the endogenous WAS promoter to drive WAS cDNA expression. There were seven patients treated in France and England, and at the time of last reported follow-up, six of seven patients were alive with no severe bleeding episodes, decreased hospitalization days, and resolution of eczema and predisposition for infections. One patient died 7 months after treatment due to preexisting, refractory herpes virus infections [33•]. In Italy, three patients have been treated, all of whom have stable engraftment of genetically corrected cells as well as improved thrombocytopenia, eczema, and infectious episodes [34•]. Importantly, molecular analyses of all nine surviving patients have demonstrated multilineage and polyclonal engraftment over time without leukemic events. Both these trials remain open in addition to a third in the USA, which has enrolled two patients who are reported to have improvement in immune and hematologic parameters without genotoxicity at early time points.
The Future of Gene Therapy
There have been tremendous improvements in the field of gene therapy for PID, even beyond what has already been reviewed. As diagnostic tools have become more sophisticated, they have allowed clinicians to better understand the pathology behind complicated clinical manifestations of disease. The growing availability of newborn screening for SCID and early diagnosis of other monogenic PID have allowed younger infants without HLA-matched donors to receive genetically corrected autologous stem cells at a time when they are more likely to successfully engraft. Always with safety as the priority, there continues to be significant progress in vector design to decrease the risk of oncogenesis and maintain high and stable transgene expression. Understanding of the complex HSC biology is growing, allowing investigators to selectively treat true stem cells most capable of self-renewal and reconstituting the entire hematopoietic lineage. There have also been improvements in cell culture conditions through co-culturing with recombinant extracellular matrix proteins and the addition of various cytokines and ligands, and there are ongoing efforts to expand human HSCs ex vivo through chemical compounds [35]. In addition, many are working to scale up the production of gene modified products in order to bring this therapeutic to a greater number of those in need.
While traditional forms of gene therapy have focused on the delivery and incorporation of functional genes to diseased cells without targeting them to their natural location in the genome, there has been an emphasis on site-specific gene therapy in recent years. The use of engineered genome editing tools such as transcription activator-like effector nucleases (TALENs), clustered regularly interspaced short palindromic repeats (CRISPRs), and zinc-finger nucleases (ZFNs) in preclinical settings are now transitioning to the clinic [36]. By creating a site-specific break in the DNA near the location of a known mutation, a cell’s natural repair mechanisms can be utilized to incorporate normal segments of DNA. This leaves genes in their endogenous locations under control of normal regulatory elements and decreasing the risk of insertional oncogenesis that can potentially occur with random integration of genetic material by a viral vector. Gene targeting for monogenic disorders of the immune system is being aggressively studied and applied to several forms of SCID, X-linked Hyper-IgM syndrome, X-linked agammaglobulinemia, and IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) amongst others. While these genome editing platforms still face many of the challenges involved in working with primitive HSCs such as potential genotoxicity and low efficiency, their ability to cure disease has already been shown in vitro and in animal models, and further refinement of the technology will likely lead to an increasing role in the clinic.
The field of gene therapy has advanced significantly since the first patient was treated in 1990 and continues to develop at a rapid pace. With improvements in vector design, understanding of stem cell biology and bone marrow transplant, and genome manipulation, it is foreseeable that gene therapy will become the standard of care for certain disease entities in the near future. It has already been shown to be beneficial for ADA and X-linked SCID, WAS, and XCGD, and a number of preclinical experiments have demonstrated the promise of targeted gene editing strategies. The large number of patients who have been treated thus far in clinical trials indicate that the field of gene therapy is evolving beyond experimentation to becoming a therapeutic standard.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Bousfiha A, Jeddane L, Al-Herz W, et al. The 2015 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. 2015;35:727–38. doi:10.1007/s10875-015-0198-5.
Pai S-Y, Logan BR, Griffith LM, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med. 2014;371(5):434–46. doi:10.1056/NEJMoa1401177. A major retrospective study of the factors influencing outcomes for patients with SCID treated by allogeneic transplant in multiple major centers across North America, highlighting the benefits of early diagnosis and treatment.
Gennery AR, Slatter MA, Grandin L, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010;126(3):602–610.e11. doi:10.1016/j.jaci.2010.06.015.
Blaese RM, Culver KW, Miller AD, et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995;270(5235):475–80. doi:10.1126/science.270.5235.475.
Chan B, Wara D, Bastian J, et al. Long-term efficacy of enzyme replacement therapy for adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID). Clin Immunol. 2005;117(2):133–43. doi:10.1016/j.clim.2005.07.006.
Serana F, Sottini A, Chiarini M, et al. The different extent of B and T cell immune reconstitution after hematopoietic stem cell transplantation and enzyme replacement therapies in SCID patients with adenosine deaminase deficiency. J Immunol. 2010;185(12):7713–22. doi:10.4049/jimmunol.1001770.
Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood. 2009;114(17):3524–32. doi:10.1182/blood-2009-06-189209.
Onodera M, Ariga T, Kawamura N, et al. Successful peripheral T-lymphocyte-directed gene transfer for a patient with severe combined immune deficiency caused by adenosine deaminase deficiency. Blood. 1998;91:30–6.
Kohn DB, Weinberg KI, Nolta JA, et al. Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat Med. 1995;1(10):1017–23.
Bordignon C, Mavilio F, Ferrari G, et al. Transfer of the ADA gene into bone marrow cells and peripheral blood lymphocytes for the treatment of patients affected by ADA-deficient SCID. Hum Gene Ther. 1993;4:513–20.
Hoogerbrugge P, van Beusechem V, Fischer A, et al. Bone marrow gene transfer in three patients with adenosine deaminase deficiency. Gene Ther. 1996;3(2):179–83.
Aiuti A PhD, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360(5):447–58. doi:10.1056/NEJMoa0805817. Report of the outcomes for the first 10 ADA-SCID patients treated by gene therapy using a gamma-retroviral vector and reduced intensity conditioning with a high rate of immune reconstitution.
Gaspar HB, Buckland K, Rivat C, et al. Immunological and metabolic correction after lentiviral vector mediated haematopoietic stem cell gene therapy for ADA deficiency. Mol Ther. 2014;22(Supplement 1):S106.
Candotti F, Shaw KL, Muul L, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency : clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120(18):3635–46. doi:10.1182/blood-2012-02-400937.There.
Carbonaro Sarracino D, Shaw K, Sokolic R, et al. U.S. clinical gene therapy trials for adenosine deaminase-deficienct severe combined immune deficiency (ADA-SCID). J Clin Immunol. 2014;34(S2):139–515. doi:10.1007/s10875-014-0101-9.
Aiuti A, Cassani B, Andolfi G, et al. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J Clin Invest. 2007;117(8):2233–40. doi:10.1172/JCI31666.
Pike-Overzet K, de Ridder D, Weerkamp F, et al. Gene therapy: is IL2RG oncogenic in T-cell development? Nature. 2006;443(7109):E5. doi:10.1038/nature05218. discussion E6-E7.
Sokolic R, Maric I, Kesserwan C, et al. Myeloid dysplasia and bone marrow hypocellularity in adenosine deaminase-deficient severe combined immune deficiency. Blood. 2011;118(10):2688–94. doi:10.1182/blood-2011-01-329359.
Carbonaro Sarracino DA, Jin X, Wang X, et al. Gene therapy/bone marrow transplantation in ADA-deficient mice: roles of enzyme-replacement therapy and cytoreduction. Blood. 2012;120(18):3677–87. doi:10.1182/blood-2012-02-408591.
Noguchi M, Yi H, Rosenblatt HM, et al. Interleukin-2 receptor γ chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993;73(1):147–57. doi:10.1016/0092-8674(93)90167-O.
Hacein-Bey-Abina S, Hauer J, Lim A, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363(4):355–64. doi:10.1056/NEJMoa1000164. Report of outcomes for subjects treated with a gamma-retroviral vector without any conditioning, leading to restoration of T cell immunity (although several developed T leukoprioliferative complications).
Gaspar HB, Parsley KL, Howe S, et al. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364(9452):2181–7. doi:10.1016/S0140-6736(04)17590-9.
Howe SJ, Mansour MR, Schwarzwaelder K, Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Investig. 2008;44(0):1–22. doi:10.1172/JCI35798DS1.
Hacein-Bey Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–42. doi:10.1172/JCI35700. A detailed and sophisticated molecular analysis of the mechanisms of insetrtional oncogenesis leading to T lymphoproliferation in XSCID subjects undergoing gene therapy with a "first generation" gamma-retroviral vector.
Hacein-Bey-Abina S, Pai S-Y, Gaspar HB, et al. A Modified γ-Retrovirus Vector for X-Linked Severe Combined Immunodeficiency. N Engl J Med. 2014;371(15):1407–17. doi:10.1056/NEJMoa1404588. A recent report of successful restoration of T lymphocyte immunity without leukoproliferation or clonal expansions, using a "second generation: gamma-retroviral vector lacking strong enhancer elements.
De Ravin SS, Gray JT, Throm RE, et al. False-positive HIV PCR test following ex vivo lentiviral gene transfer treatment of X-linked severe combined immunodeficiency vector. Mol Ther. 2014;22(2):244–5. doi:10.1038/mt.2013.296.
Seger RA, Gungor T, Belohradsky BH, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood. 2002;100(13):4344–50. doi:10.1182/blood-2002-02-0583. A multi-center trial of reduced intensity conditioning for allogeneic transplant of Chronic Granulomatous Disease with excellent survival, even with unrelated donors.
Kang EM, Choi U, Theobald N, et al. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood. 2010;115(4):783–91. doi:10.1182/blood-2009-05-222760. This clinical trial of gene therapoy for CGD using a gamma-retroviral vector achieved only low level engraftment of gene corrected stem cells, yet still provided some benefit to subjects, providing proof-of-principle for this approach.
Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich syndrome. 2008.
Moratto D, Giliani S, Bonfim C, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980-2009: an internationalcollaborative study. Blood. 2011;118(6):1675–85. doi:10.1182/blood-2010-11-319376.
Boztug K, Banerjee PP, Ph D, et al. Stem-cell gene therapy for the Wiskott-Aldrich Syndrome. N Engl J Med. 2010;363(20):1918–27.
Braun CJ, Boztug K, Paruzynski A, et al. Gene therapy for wiskott-Aldrich syndrome—long-term efficacy and genotoxicity. Sci Transl Med. 2014;6(227):1–14. doi:10.1126/scitranslmed.3007280.
Hacein-Bey Abina S, Gaspar HB, Blondeau J, et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich Syndrome. JAMA. 2015;313(15):1550. doi:10.1001/jama.2015.3253. Successful restoration of immunity, with partial improvement in platelet counts, by gene therapy for Wiskott-Aldrich Syndrome using a lentiviral vector.
Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich Syndrome. Science. 2013;341(6148):1–12. doi:10.1126/science.1233151. Successful restoration of immunity, with partial improvement in platelet counts, by gene therapy for Wiskott-Aldrich Syndrome using a lentiviral vector.
Csaszar E, Kirouac DC, Yu M, et al. Rapid expansion of human hematopoietic stem cells by automated control of inhibitory feedback signaling. Cell Stem Cell. 2012;10(2):218–29. doi:10.1016/j.stem.2012.01.003.
Pu J, Frescas D, Zhang B, Feng J. Utilization of TALEN and CRISPR/Cas9 technologies for gene targeting and modification. Exp Biol Med. 2015;240(8):1065–70. doi:10.1177/1535370215584932.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Drs. Kuo and Kohn declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Immune Deficiency and Dysregulation
Rights and permissions
About this article

Cite this article
Kuo, C.Y., Kohn, D.B. Gene Therapy for the Treatment of Primary Immune Deficiencies. Curr Allergy Asthma Rep 16, 39 (2016). https://doi.org/10.1007/s11882-016-0615-8
Published:
DOI: https://doi.org/10.1007/s11882-016-0615-8