
Abstract
We report on our research in using literature-based discovery (LBD) to provide pharmacological and/or pharmacogenomic explanations for reported adverse drug effects. The goal of LBD is to generate novel and potentially useful hypotheses by analyzing the scientific literature and optionally some additional resources. Our assumption is that drugs have effects on some genes or proteins and that these genes or proteins are associated with the observed adverse effects. Therefore, by using LBD we try to find genes or proteins that link the drugs with the reported adverse effects. These genes or proteins can be used to provide insight into the processes causing the adverse effects. Initial results show that our method has the potential to assist in explaining reported adverse drug effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adverse drug effects pose significant health and financial problem worldwide. The World Health Organization (WHO) defines “pharmacovigilance” as “the science and activities relating to detection, assessment, understanding, and prevention of adverse effects or any other drug related problems”. Some of the adverse effects are detected during clinical trials, but some are detected after the drugs come to market. Considerable research and effort in pharmacovigilance is dedicated to adverse drug effect signal detection. Most often spontaneous reporting systems (SRSs) such as FAERS are used for detecting signals with statistical and data mining algorithms [1]. Adverse drug effects can also be detected in bibliographic databases such as MEDLINE [2]. Electronic patient records are another resource for detection of adverse drug effects [3]. Sometimes, combined signals from various sources can be used for adverse drug effects detection [4]. Recently, social media, such as medical message boards [5] and Twitter [6] has been used for adverse drug effect detection.
In contrast to the majority of other pharmacovigilance methods, whose goal is to detect drug safety signals, our goal is to provide an explanation for known adverse drug effects. More specifically, our goal is to provide a pharmacological and/or pharmacogenomics explanation by finding genes or proteins that link the drug to the observed adverse effect. Our basic assumption is that the drugs have some effect on some genes or proteins and that these genes or proteins are associated with the observed adverse effects.
Methods
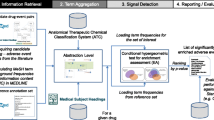
We use Literature-based Discovery (LBD) [7] to find explanations for (drug, adverse effect) pairs. The goal of LBD is to generate novel hypotheses by analyzing the literature and optionally other knowledge sources. LBD uses either of two basic approaches: open discovery and closed discovery; both are based on a paradigm of three related concepts: X, Y, and Z. In open discovery only the starting concept is known. For example, if we want to find a new treatment for a given disease (X), we first try to find (patho)physiological characteristics (Y) of the disease and then seek drugs (Z) that can deal with these characteristics. In closed discovery both the starting concept (X) and the end concept (Z) are known, and we want to find intermediate, linking concepts (Y) that may help explain the relationship between X and Z. In any case, LBD is meant as a discovery support paradigm. LBD generates hypotheses, but a knowledgeable human expert is needed for the interpretation of these hypotheses [8]. Our methodology is meant to assist an experienced pharmacovigilance expert.
For the current study closed discovery is better suited because we work with known adverse effects. In other words, the starting concept (Drug_X) is known as well as the end concept (Adverse_effect_Z), and we want to find Genes_or_proteins_Y that somehow link the drug with the adverse effects. By finding the linking genes or proteins, we provide an explanation for an association found statistically.
For this research we used the closed discovery component of a LBD tool called SemBT [9, 10] available at [11]. SemBT uses semantic relations extracted with the SemRep [12] natural language processing system from all of MEDLINE.
Results
The SemBT version used for this study is based on semantic relations extracted with SemRep from 44,250,865 sentences. These sentences come from 23,657,386 MEDLINE citations (the entire MEDLINE database up to the end of March 2014). 15,175,993 distinct semantic relations were extracted from a total of 69,331,058 semantic relation instances.
Statistical evaluation
To evaluate our methodology we selected 51 true positive and 29 true negative (drug, adverse effect) pairs that were curated by pharmacovigilance experts. All the 29 true negatives and 28 of the true positives came from the EU-ADR project [2] because it is a well-established benchmark used in several recent pharmacovigilance papers. The additional 23 true positive pairs were added by pharmacovigilance experts because they believed that these pairs likely had pharmacogenomic explanation. For each pair, we created a ranked list of linking Y genes or proteins using SemBT.
For the group of 51 true positive pairs, we found a total of 1523 linking Y genes or proteins, giving 29.86 Ys per true positive pair. For the group of 29 true negative pairs, we found a total of 392 linking Ys, giving 13.52 Ys per true negative pair. The Nonparametric Mann–Whitney test for comparison of two independent samples was used to compare the number of Ys found in the two groups. There was a significant difference between the groups (p = 0.00975), with Group 1 having significantly higher values than Group 2. Therefore, our method finds considerably more Ys per (drug, adverse effect) pair for the true positive pairs than for the true negative pairs. For us this is an indication that our basic idea is valid, i.e. explaining adverse drug effects through the genes and/or proteins that link the drug to the disease.
Potentially new adverse drug effect explanations
For each true positive (drug, adverse effect) pair, a ranked list of linking genes or proteins was produced and given to a pharmacologist for expert evaluation. The linking genes or proteins (Ys) were ranked by the sum of distinct relations between the drug (X) and the Ys plus the distinct relations between the Ys and the adverse effect (Z). The pharmacologist found out that in the majority of cases, the adverse effect was due to the drug’s primary pharmacological effect, i.e. drug’s major mechanism of action, as was expected. However, he found a considerable number of cases where the adverse effect was not caused by the major drug action and therefore represented potentially novel ways to explain the adverse drug effect. Some of these cases are shown in Table 1.
The examples in the table are explained in more detail below.
Azathioprine
Azathioprine is an immunosuppressive drug that is metabolized to 6-mercaptopurine, a purine analogue that inhibits DNA synthesis by inhibiting the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT). This leads to a cytotoxic effect in dividing cells; therefore, some of the reported adverse side effects, such as leucopenia, cytopenia, myelosuppression, and anemia, can be explained by the main mechanism of action. However, here we provide novel LBD approach to identify the protein targets to explain other reported adverse side effects, in particular, acute pancreatitis and hepatotoxicity.
To provide a new hypothesis for the mechanism of azathioprine-induced acute pancreatitis, we identified pancreatic lipase and glutathione S-transferase as protein targets, as shown in Table 1. Application of azathioprine can lead to an asymptomatic increase in pancreatic enzymes, such as lipase and amylase [13]. Indeed, the onset of acute pancreatitis is positively correlated with the abnormally high pancreatic enzyme levels, e.g. pancreatic lipase and amylase [14]. Importantly, pancreatic lipase is the key enzyme in the development of acute pancreatitis by releasing membrane-toxic fatty acids [15]. Moreover, azathioprine is a competitive inhibitor of glutathione S-transferase [16], and can thus lead to glutathione (GSH) depletion. Since GSH is an important intracellular antioxidant this leads to increased cellular oxidative stress. Indeed, GSH depletion is correlated with the pancreatitis [17]. Furthermore, hepatotoxicity is also correlated with GSH depletion [18], which was also detected by our SemBT software.
Irinotecan
Irinotecan is bioactivated by carboxylesterases to SN-38, a molecule which is an inhibitor of topoisomerase I, and thus leads to the inhibition of both DNA replication and transcription in dividing cells. Thus, some of the reported adverse side effects, such as myelosuppression, neutropenia, and cytopenia, can be explained directly by the cytotoxic action (main mechanism of drug action) on dividing immune cells [19]. However, to explain other common adverse reactions, such as diarrhea, we applied literature-based discovery for identifying target proteins, as presented with semantic relations in Table 1. To explain diarrhea, we identified the uridine diphosphate glucoronosyltransferase 1A1 (UGT1A1) as a target protein. UGT1A1 is involved in the inactivation of the bioactive molecule SN-38 by glucuronidation [19]. Indeed, patients bearing certain specific gene polymorphisms of UGT1A1 have a higher risk of severe neutropenia and diarrhea [20].
Atorvastatin, simvastatin, pravastatin
Although statins are well tolerated in most patients, around 7–29 % of them have statin-associated muscle symptoms [21], which are now recognized as a clinically significant complication of statin therapy. There is a knowledge gap in understanding the mechanism of statin-induced rhabdomyolysis, and even more in their therapy. Thus, we tried to use the literature-based discovery approach to identify the target proteins, which might explain these statin-associated muscle side-effects. We used atorvastatin, simvastatin, and pravastatin as representative drugs of statins, and identified the SLCO1B1 gene encoding the OATP1B1 protein, Carnitine O-Palmitoyltransferase, and Cytochrome P450 3A4 as target proteins involved in statin-induced rhabdomyolysis. The semantic relations identified are presented in Table 1.
The first target was OATP1B1, which belongs to the family of a solute carrier organic anion transporters, and is an influx membrane transporter responsible for the uptake of statins into hepatocytes. Changes in its activity, either by drug-drug interactions or by SLCO1B1 gene polymorphism, can affect the pharmacokinetics of statins [22]. For example, the inhibition, or lower activity, can lead to increased bioavailability (higher plasma concentrations of statins), and thus to adverse reactions, such as rhabdomyolysis. The second target identified was Carnitine O-Palmitoyltransferase (CPT), which is a mitochondrial transferase enzyme involved in the metabolism of palmitoylcarnitine into palmitoyl-CoA. Abnormal regulation of CPT can cause rhabdomyolysis [23]. Importantly, statins can interfere with CPT activity, e.g. in one study atorvastatin increased the expression of CPT [24]. Moreover, CPT deficiency often also causes non-exercise-induced rhabdomyolysis [25]. The third target identified was Cytochrome P450 3A4 (CYP3A4), which is one of the most important enzymes involved in the drug metabolism. Importantly, statins are metabolized by CYP3A4, as they also inhibit its activity [26]. Therefore, concomitant administration of statin therapy and drugs that inhibit CYP3A4 increases the risk of rhabdomyolysis [27].
Semantic relation extraction evaluation
The quality of the explanations for the drug adverse effects provided in our approach largely depends on the quality of the semantic relation extraction process. Therefore, we conducted an evaluation to estimate the accuracy of the semantic processing. The evaluation was conducted at the semantic relation instance level. In other words, the goal was to determine whether a particular semantic relation was correctly extracted from a particular sentence. Eighty subjects, students in the final year of medical school (Faculty of Medicine, University of Maribor) received intensive training and detailed instructions on how to evaluate before conducting the evaluation. Subjects were organized in such a way that three of them independently evaluated the same semantic relation instance. However, subjects could decide whether to skip a relation to be evaluated and which ones to evaluate from the set of assigned relations. Therefore, it turned out that although most of the instances were evaluated by three subjects, not all were.
The semantic relation instances evaluated were a subset of those relevant to the true positive and true negative adverse drug effects mentioned before. In total 4069 semantic relation instances were evaluated 10,279 times. The instances were evaluated as correct 8646 times (84 %) and as incorrect 1633 times (16 %). 3795 distinct instances were evaluated as correct (93 %) at least once and 1068 distinct instances were evaluated as incorrect (26 %) at least once. If we did not take into account the number of persons who evaluated a particular relation instance, we found that 3369 (82 %) distinct instances were evaluated more frequently as correct than as incorrect: 442 (11 %) instances were evaluated more often as incorrect than as correct, and 258 (7 %) relation instances were evaluated as correct exactly as many times as they were evaluated as incorrect. However, if we consider only the relation instances being evaluated by exactly three evaluators (N = 1500), then 1321 (88 %) relation instances were evaluated more times as correct than as incorrect, and 179 (12 %) instances were evaluated more times as incorrect than as correct, 1062 instances were always evaluated as correct (71 %) and 45 distinct instances were always evaluated as incorrect (3 %).
Conclusions
We presented a tool and a methodology for finding pharmacological and/or pharmacogenomics explanations for known adverse drug effects through genes or proteins that link the drugs to the adverse effects. We found several potentially novel explanations, which cannot be explained by the drug’s major mechanism of action.
References
Sakaeda, T., Tamon, A., Kadoyama, K., and Okuno, Y., Data mining of the public version of the FDA adverse event reporting system. Int. J. Med. Sci. 10:796–803, 2013. doi:10.7150/ijms.6048.
Avillach, P., Dufour, J.-C., Diallo, G., et al., Design and validation of an automated method to detect known adverse drug reactions in MEDLINE: a contribution from the EU-ADR project. J. Am. Med. Inform. Assoc. 20:446–52, 2013. doi:10.1136/amiajnl-2012-001083.
Warrer, P., Hansen, E. H., Juhl-Jensen, L., and Aagaard, L., Using text-mining techniques in electronic patient records to identify ADRs from medicine use. Br. J. Clin. Pharmacol. 73:674–84, 2012. doi:10.1111/j.1365-2125.2011.04153.x.
Li, Y., Ryan, P. B., Wei, Y., and Friedman, C., A method to combine signals from spontaneous reporting systems and observational healthcare data to detect adverse drug reactions. Drug Saf. 38:895–908, 2015. doi:10.1007/s40264-015-0314-8.
Benton, A., Ungar, L., Hill, S., et al., Identifying potential adverse effects using the web: a new approach to medical hypothesis generation. J. Biomed. Inform. 44:989–96, 2011. doi:10.1016/j.jbi.2011.07.005.
Freifeld, C. C., Brownstein, J. S., Menone, C. M., et al., Digital drug safety surveillance: monitoring pharmaceutical products in twitter. Drug Saf. 37:343–350, 2014. doi:10.1007/s40264-014-0155-x.
Swanson, D. R., Fish oil, Raynaud’s syndrome, and undiscovered public knowledge. Perspect. Biol. Med. 30:7–18, 1986.
Hristovski, D., Rindflesch, T., and Peterlin, B., Using literature-based discovery to identify novel therapeutic approaches. Cardiovasc. Hematol. Agents Med. Chem. 11:14–24, 2013.
Hristovski, D., Kastrin, A., Peterlin, B., and Rindflesch, T. C., Combining Semantic Relations and DNA Microarray Data for Novel Hypotheses Generation. Link Lit. Inf. Knowl. Biol. 6004(Str):53–61, 2010. doi:10.1007/978-3-642-13131-8.
Hristovski, D., Dinevski, D., Kastrin, A., and Rindflesch, T. C., Biomedical question answering using semantic relations. BMC Bioinform. 16:6, 2015. doi:10.1186/s12859-014-0365-3.
Hristovski D., SemBT. http://sembt.mf.uni-lj.si. 2009.
Rindflesch, T. C., and Fiszman, M., The interaction of domain knowledge and linguistic structure in natural language processing: interpreting hypernymic propositions in biomedical text. J. Biomed. Inform. 36:462–77, 2003. doi:10.1016/j.jbi.2003.11.003.
Liverani, E., Leonardi, F., Castellani, L., et al., Asymptomatic and persistent elevation of pancreatic enzymes in an ulcerative colitis patient. Case Rep. Gastrointest. Med. 2013:415619, 2013. doi:10.1155/2013/415619.
Ventrucci, M., Pezzilli, R., Naldoni, P., et al., Serum pancreatic enzyme behavior during the course of acute pancreatitis. Pancreas 2:506–9, 1987.
Schmitz-Moormann, P., Comparative radiological and morphological study of the human pancreas. IV. acute necrotizing pancreatitis in man. Pathol. Res. Pract. 171:325–35, 1981. doi:10.1016/S0344-0338(81)80105-7.
Magos, L., Cikrt, M., and Snowden, R., The dependence of biliary methylmercury secretion on liver GSH and ligandin. Biochem. Pharmacol. 34:301–5, 1985.
Schoenberg, M. H., Büchler, M., Pietrzyk, C., et al., Lipid peroxidation and glutathione metabolism in chronic pancreatitis. Pancreas 10:36–43, 1995.
Akai, S., Hosomi, H., Minami, K., et al., Knock down of gamma-glutamylcysteine synthetase in rat causes acetaminophen-induced hepatotoxicity. J. Biol. Chem. 282:23996–4003, 2007. doi:10.1074/jbc.M702819200.
Kuhn, J. G., Pharmacology of irinotecan. Oncology (Williston Park) 12:39–42, 1998.
Xu, J.-M., Wang, Y., Ge, F.-J., et al., Severe irinotecan-induced toxicity in a patient with UGT1A1 28 and UGT1A1 6 polymorphisms. World J. Gastroenterol. 19:3899–903, 2013. doi:10.3748/wjg.v19.i24.3899.
Stock, J., Statin-associated muscle symptoms EAS Consensus Panel paper focuses on this neglected patient group. Atherosclerosis 242:346–50, 2015. doi:10.1016/j.atherosclerosis.2015.06.049.
Niemi, M., Transporter pharmacogenetics and statin toxicity. Clin. Pharmacol. Ther. 87:130–3, 2010. doi:10.1038/clpt.2009.197.
Schröder, J. P., Mau, W., Schumacher, S., and Zierz, S., Abnormal regulation of carnitine palmitoyltransferase in monozygotic twins as the cause of rhabdomyolysis. Dtsch. Med. Wochenschr. 115:337–9, 1990. doi:10.1055/s-2008-1065012.
Roglans, N., Sanguino, E., Peris, C., et al., Atorvastatin treatment induced peroxisome proliferator-activated receptor alpha expression and decreased plasma nonesterified fatty acids and liver triglyceride in fructose-fed rats. J. Pharmacol. Exp. Ther. 302:232–9, 2002.
Keverline, J. P., Recurrent rhabdomyolysis associated with influenza-like illness in a weight-lifter. J. Sports Med. Phys. Fitness 38:177–9, 1998.
Yang, S.-H., Choi, J.-S., and Choi, D.-H., Effects of HMG-CoA reductase inhibitors on the pharmacokinetics of losartan and its main metabolite EXP-3174 in rats: possible role of CYP3A4 and P-gp inhibition by HMG-CoA reductase inhibitors. Pharmacology 88:1–9, 2011. doi:10.1159/000328773.
Dopazo, C., Bilbao, I., Lázaro, J. L., et al., Severe rhabdomyolysis and acute renal failure secondary to concomitant use of simvastatin with rapamycin plus tacrolimus in liver transplant patient. Transplant. Proc. 41:1021–4, 2009. doi:10.1016/j.transproceed.2009.02.019.
Acknowledgments
This work was supported in part by the Intramural Research Program of the U.S. National Institutes of Health, National Library of Medicine. Authors would like to thank Celine Narjoz and Marie-Anne Loriot for suggesting the additional adverse drug reactions, which we used in this study. We are also grateful for the contribution of the medical students (Faculty of Medicine, University of Maribor) in the evaluation of the extracted relations.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Education & Training.
Rights and permissions
About this article

Cite this article
Hristovski, D., Kastrin, A., Dinevski, D. et al. Using Literature-Based Discovery to Explain Adverse Drug Effects. J Med Syst 40, 185 (2016). https://doi.org/10.1007/s10916-016-0544-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10916-016-0544-z