
Abstract
Dehydration of wet bacterial cellulose (BC) mainly focuses on the physical characteristic or mechanical strength, but its polymorph crystal structure and thermal properties were rarely discussed. Herein, this study utilizes the drying method of hot-pressed and freeze-dried to the wet BC to elucidate the BC polymorph crystal structure in correlation with the BC crystallinity and thermal properties. The results indicate that dehydration of BC using hot-pressed (BC-HP) method reduced the cellulose Iα allomorph fraction by ± 5.0%, increased the crystallinity by ± 5.0%, but lower in thermal stability compared to freeze-dried (BC-FD) method. Additionally, BC-HP shows an exothermic reaction of melting at lower enthalpy value which is contradict to BC-FD and plant cellulose that presented an endothermic reaction with higher enthalpy values. Thus, investigation on BC polymorph crystal structure and thermal properties may offer an important role to regulate the desired characteristic of the dried BC.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recently, the utilization of agro-industrial wastes as an alternative fermentation medium in bacterial cellulose (BC) production has been explored extensively due to the limitation of the current conventional medium such as HS medium and coconut water medium that required expensive investment for the carbon source and nutrients [1]. The capability of the alternative mediums diverges greatly in terms of nutritional quality and quantity, thus leading to a variation in BC optimum production [2]. Meanwhile, the nature of the BC produced varies significantly depending on the type of bacteria used for cultivation and the chemical components and compositions of selected medium [3]. Furthermore, the post-processing procedures for BC cleansing including the removal of cell body mass and medium residue by using chemical treatment such NaOH could further altered the characteristic of BC considerably [4].
Purified BC as the final product can be accepted in the form of wet or dry state of pellicles, homogenous fibrous slurry, dried thin films, etc. Normally, the application of BC in industry will determined the final form of the BC to be delivered. For example, a product of Nata de Coco contained of small cube of wet BC pellicles, while tailored form of wet BC pellicles is applied as an artificial skin for wound healing treatment, and dried thin films are used as an acoustic element [5,6,7]. On top of that, for the application of BC as an intermediate material especially in drug delivery and food additives, a dry state form is preferable for easier in handling, storage, and transportation of the final product [8].
Drying of wet BC can be achieved by dehydration process commonly using either thermo-mechanical method such as hot-pressed or cold method such as freeze-dried [9]. The drying procedures are the key factors to the distinctions in BC properties and characteristic. Previous studies were mainly focusing on the effect of drying regarding the morphology, water holding capacity, mechanical properties, and crystallinity of the dried BC. For instance, the BC nanofibril appeared in a densely layered structure by using air-drying method, while freeze-drying method leads to a complex microporous network [10, 11]. However, despite the afore-mentioned studies of the dried BC properties and applications, information and research of the effect of drying method to the BC polymorph crystalline structure and its correlation to the crystallinity and thermal properties are lacking. Hence, investigation on this matter may offer a new perspective to further elucidate the dried BC properties and characteristic.
Therefore, this study aims to investigate the effect of drying method applied to the wet BC pellicles newly produced from OPF juice medium in association with cellulose allomorph crystalline structure and its correlation to the BC crystallinity, crystallite size, and thermal properties. The works presented include depth analysis using FTIR, XRD, TGA, and DSC for the dried BC samples from hot-pressed method and freeze-dried method. Moreover, a systematic comparison using α-cellulose and MCC from plant-based cellulose is also carried out.
Material and method
Materials
Fresh OPF petioles were pressed using pilot scale hydraulic pressing machine (Matsuo Inc., Japan) for juice extraction [12] and stored at − 18 °C up until usage. Sodium hydroxide pellets (NaOH, ACS reagent, R&M Chemicals) and glacial acetic acid (CH3CO2H, A.R., R&M Chemicals), α-cellulose (Sigma-Aldrich (M)), and microcrystalline cellulose (MCC, Sigma-Aldrich (M)) were used as received. The NaOH pellets were dissolved using distilled water and further diluted to 0.5 N solution, while the acetic acid was diluted using distilled water to 50% v/v concentration. Acetobacter xylinum 0416 liquid inoculum was obtained from Malaysian Agricultural Research and Development Institute (MARDI) and used as received.
BC production
OPF juice was defrosted and vacuum filtered through a cotton filter cloth to remove solid particle residues from the juice. The culture medium was prepared in 250-ml conical flask containing 100 ml of OPF juice that was diluted to 60 v/v% concentration by using distilled water, and the pH medium was adjusted to pH 4.0 using acetic acid 50% (v/v) aqueous solution [13]. For sterilization procedure, the media were autoclaved for 10 min at 121 °C.
The starter liquid inoculum was added at 10% v/v into the medium aseptically and incubated statically for at least 14 days at 30 °C for a maximum BC production. The BC pellicles obtained from the incubation process were washed and then treated with 0.5 N NaOH solution at 80 °C for 1 h for cell mass and medium residue removal. Further purification on the BC pellicles was done by immersing in 5% (v/v) NaOCl solution to completely remove NaOH residue for 12 h followed by thorough washing and rinsing until neutralized.
BC dehydration
The purified BC pellicles were separated into two types of drying. First, the BC pellicles were placed in between filter papers and hot-pressed at 80 °C for 10 min and air-dried until constant weight and the sample was noted as BC-HP. Secondly, the BC pellicles were placed on polystyrene Petri dish and freeze-dried at − 80 °C for 48 h and the sample was noted as BC-FD. The morphology of BC-FD and BC-HP was examined using field emission scanning electron microscope (FE-SEM) JEOL JSM 7800 F with an accelerating voltage of 5–15 kV. The surface of the sample was coated with platinum using JEOL JFC-1600 Auto Fine Coater at 5 kV.
FTIR
A chemical structure of the α-cellulose, MCC, BC-FD, and BC-HP sample was analyzed using FTIR spectrometry (Thermo Scientific Co., Nicolet iS5) based on attenuated total reflection (ATR) data ranging over 4000–400 cm−1. The spectra were recorded with an accumulation of 256 scans at 4 cm−1 resolutions in the absorbance mode.
Further analysis has been done from the data obtained which were the crystallinity index (\(CrI\)) [14] and the cellulose \(I\) α fraction [15]. The value of \(CrI\) has been approximated by using the absorption ratio as follows (Eq. 1) [14].
where A1420 and A897—absorption value at 1420 cm−1 and 897 cm−1.
Meanwhile, the fraction of cellulose \(I\)α (\(f_{ir}\)) was specified from the following Eq. (2) [15]:
The band area of \(I\)α at 750 cm−1 (A750) and \(I\)β at 710 cm−1 (A710) was obtained by peak deconvolution using Gaussian–Lorentzian curve fitting analysis.
XRD
The cellulose crystal structure of the α-cellulose, MCC, BC-FD, and BC-HP sample was observed using Bruker D8 Advanced X-ray Diffractometer (XRD) with Ni-filtered CuKα radiation (λ = 0.154056 nm). The XRD was operated at 45 kV and 40 mA. The samples were scanned covering 2θ angle range from 5° to 60° at a rate of 2° per minutes.
The value of crystallinity index (CrI) for each sample was evaluated between the height of the maximum intensity of crystalline fraction and intensity of amorphous fraction without baseline subtraction as described by Segal method and presented in the following equation (Eq. 3) [16]:
Additionally, the value for crystallinity (Cr) was also calculated by peak deconvolution method using Origin software. Each possible crystalline peak was deconvoluted using Gaussian function after baseline subtraction to separate the amorphous and crystalline diffraction spectrum. Then, Cr was determined by the ratio of the total area of crystalline peaks to the total area of the spectra from 10° to 40° 2θ as the baseline using the following equation (Eq. 4) [17]:
The interplanar spacing (d-spacing) of respected plane was determined by the Bragg’s Eq. (5) [18]:
where λ is the X-ray wavelength at 0.154056 nm, and θ is the Bragg angle.
Furthermore, the Scherrer formula was employed to calculate the crystallite size (CS) for each sample as follows (Eq. 6)[19]:
herein λ indicated the X-ray wavelength at 0.154056 nm, k is the value for Scherrer constant at 0.9, β is referred as the full-width at half peak of D in radian, and θ is the Bragg angle.
The monoclinic and triclinic relation (Iα/Iβ) was estimated using the function established by Wada et al. [20] from the discriminant analysis of two equatorial d-spacings (d1 and d2) as shown in equation (Eq. 7)[20]:
where d1 and d2 were obtained from the calculation of d-spacing at peak 1 (100Iα or 110Iβ) and peak 2 (110Iβ or 010Iα) respectively. Herein, for Z > 0 specifies the cellulose sample is Iα-rich type, while Z < 0 specifies for Iβ-dominant type.
TGA and DSC
The dynamic weight loss and thermal decomposition behavior of the α-cellulose, MCC, BC-FD, and BC-HP sample were analyzed by operating a thermogravimetric analyzer (TGA 2000, TA Instruments, USA). The samples were heated on an alumina sampling pan from 25 to 600 °C at a rate of 10 °C/min under the nitrogen (N2) gas purging with a flow rate of 40 mL/min.
The heat flow analysis of the samples was measured and analyzed by operating a differential scanning calorimeter (DSC Q2000, TA Instruments, USA). The samples were weight around 5.0 mg in an aluminum sample pan and sealed with lid using encapsulating press. The heat flow measurement was recorded from − 50 to 550 °C at a rate of 10 K/min in N2 gas flow of 40 mL/min under N2 atmosphere at 60 mL/min with an empty aluminum-sealed pan used as the reference.
Results and discussion
Bacterial cellulose pellicles
Figure 1a shows the BC pellicles produced by Acetobacter xylinum 0416 cultivated using OPF juice medium. From Fig. 1b, the purified wet BC pellicles visualized as a white semi-transparent pellicle with a soft gel-like structure. This appearance of the BC pellicles was attributed to the high-water loading capacity of the BC nanofibrils structure synthesized by the bacterial. However, the physical appearance and characteristic of the BC pellicles significantly changed as dehydration took place upon drying. From Fig. 1c, the appearance of BC-HP resembled a plastic-like semi-transparent thin sheet film upon drying by using the hot-pressed unit. This phenomenon can be related to the thermo-mechanical force applied to the BC pellicles that vaporized the water molecules via in situ heat and pressing, thus pilling the nanofibrillated structure densely as shown in Fig. 1e [9]. Meanwhile, from Fig. 1d, as the result of applying the freeze-drying method, the BC-FD appeared to be a milky white with sponge-like structure. This result suggested that thru freeze-drying, while maintaining the shape of the pellicle, the water forcibly removed at instant thus leaving the microporous structure unattended in the BC pellicles as shown in Fig. 1f [10]. Overall, in regard to the unique physical appearances possessed by BC-FD and BC-HP, detail analysis was carried out microstructure level to emphasize the significant of drying method for further application.

Physical appearances of BC pellicles produced from OPF juice medium. a BC pellicles after 14 days of cultivation period, b purified wet BC pellicles, c hot-pressed drying applied to the BC pellicles, d freeze-dried drying applied to the BC pellicles, e BC-HP surface view at 30 k× magnification, f BC-FD surface view at 30 k× magnification
FTIR analysis
The FTIR spectra of the bacterial cellulose produced are illustrated in Fig. 2, along with α-cellulose and MCC as a comparison. The BC-HP and BC-FD presented almost comparable in characteristic peaks found in α-cellulose and MCC with either slightly amplified in intensity or shifted to increase or decrease in wave number. These variances suggest that biosynthesis of cellulose by bacteria has a different arrangement in cellulose structure compared to plant cellulose, but the chemical structure of cellulose remains unchanged [21]. Furthermore, FTIR spectra for BC-HP and BC-FD were nearly identical in terms of peak appearances, which suggest that drying process (heating to 80 °C nor freeze-dried at − 80 °C) process did not altered the cellulose chemical structure in BC. However, the intensity of the BC-HP spectra was higher than BC-FD with variances in peak characteristic.

FTIR spectra of cellulose samples. a IR spectra for functional group determination and analysis on degree of crystallinity. b and c IR spectra analysis for cellulose allomorphs
The cellulose crystalline form and amorphous form can be elucidated by the absorbance band at 1430 cm−1 (crystalline) and 897 cm−1 (amorphous). In addition, peak characteristic at these bands is able to give an insight on the degree of cellulose crystallinity relatively. Hence, the cellulose crystallinity index (CrI-IR) was calculated using Eq. (2) by the ratio of the absorbance value at 1430 cm−1 and 897 cm−1 [15] as shown in Table 1. BC-HP was observed to have the highest value of CrI, followed by BC-FD, MCC, and α-cellulose. This trend indicated that BC has higher crystallinity compared to other cellulose sources that were consistent with other researches cited in the literature [17]. On the other hand, the peak at 1430 cm−1 for α-cellulose is observed to have broader band characteristic compared to others which appeared to have sharp band characteristic. In the meantime, at 897 cm−1 band, a sharp and higher in intensity band for α-cellulose is observed, while other samples show lower in intensity and broader band characteristic. These phenomena are related to the degree of crystallinity in cellulose which suggested that the crystallinity increases as the band at 1430 cm−1 became sharper and more distinct, while at 897 cm−1, the band became broader and less distinct or conversely vice versa as the crystallinity decreases [22]. However, CrI obtained from FTIR data is limited only in relative value (higher or lower) that can be used conveniently for qualitative comparison [17], but other method assessing the CrI, mainly XRD, is expected to give the most realistic value [23].
Figure 2b, c show FTIR spectra at around O–H stretching group and polymorph crystalline peak of cellulose Iα (triclinic) and Iβ (monoclinic). According to Sugiyama et al. [24], the absorbance bands at 3220 cm−1 and 750 cm−1 assigned for the cellulose Iα allomorph, whereas the absorbance band at 3280 cm−1 and 710 cm−1 accounts for the cellulose Iβ allomorph. BC produced in this study evidently showed that it contained polymorph of Iα and Iβ based on this peak’s visibility. However, the appearance of peak 3220 cm−1 was shifted to 3230 cm−1 and the peak of 750 cm−1 was shifted to 748 cm−1 for BC-FD and BC-HP. Meanwhile, the appearance of peak 3280 cm−1 was shifted to 3330 cm−1 and the peak of 710 cm−1 was shifted to 711 cm−1 for BC-FD and BC-HP. It is noteworthy to mention that the peaks for Iα allomorph at BC-HP spectra have a lower and broader range compared to BC-FD and thus suggested that BC-HP has lower amount of Iα allomorph. Instead, for the α-cellulose and MCC, both were dominated mainly by the cellulose Iβ allomorph based on peaks disappearance at 3230 cm−1 and 750 cm−1. Further analysis was carried out to evaluate the fraction of cellulose Iα to cellulose Iβ allomorph as listed in Table 1. The result shows that the value of Iα fraction (fir) in BC-FD was slightly higher than BC-HP and hence confirmed the afore-mentioned on the peak characteristic differences. This result also suggested that heating process for drying applied to BC-HP has changing some of the fractions of metastable cellulose Iα to a stable cellulose Iβ structure [25]. Meanwhile, the Iα fraction value for α-cellulose and MCC was not able to be estimated due to disappearance of the peak at 3220 cm−1 and 750 cm−1 respectively. The peak disappearance can be interpreted by the relationship between O–H stretching groups and allomorph peaks. Furthermore, peak desertion at around 3240 cm−1 indicates a lower content of Iα allomorph and suggests that the inter-molecular bonds are weaker and low in crystallinity [26, 27].
XRD analysis
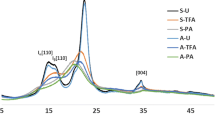
Figure 3 illustrates the crystalline structure of BC-FD, BC-HP, α-cellulose, and MCC. The XRD patterns correspond to the Miller indices, the invariant peaks of [(100), (010), (110)] crystallographic planes of cellulose Iα orientation for BC-HP and BC-FD [28] and, meanwhile, the peaks of [(110), (1–10), (200), (004)] crystallographic planes of cellulose Iβ orientation for α-cellulose and MCC [28]. Moreover, the XRD patterns for BC, α-cellulose, and MCC were consistent with other works [29, 30]. However, BC-HP represents relatively higher-intensity values of peaks compared to BC-FD, indicating a possibility of the nature sample’s physical property related to the drying or higher degree in cellulose crystallinity. It has been reported that the peaks intensity was affected by the sample particle size, which the powdered sample has a lower-intensity value compared to solid sample [31]. It is noteworthy that the BC-HP thin-film sample was not subjected to pulverize as the attempted grinding process using various types of grinding machines has utterly damaging the samples. On the other hand, for the (010) plane, BC-HP and BC-FD shared similar pattern of XRD spectra, while the α-cellulose and MCC represent comparable XRD pattern to each other with shoulder peak at (110) plane. Additionally, it is observed that XRD spectra for α-cellulose and MCC exhibited a sharp distinct peak at 34.74° and 34.64°, respectively, which assigned to (004) plane compared to BC-FD and BC-HP that exhibit a weak broad peak at this point. According to the report by Tyagi et al. [32], diffraction peak at 34.5° of wood cellulose was attributed to Iβ phase and this suggests that α-cellulose and MCC possessed a higher content of cellulose Iβ.

XRD patterns for the spectra of cellulose samples. a BC-HP and BC-FD XRD spectra and b α-cellulose and MCC XRD spectra
Table 2 summarizes the principal peaks observed at 2θ, the difference in Bragg angle, and the result obtained for the calculation of crystallinity index (CrI) and crystallinity (Cr). For the principal peaks’ observation, the BC-HP and BC-FD have a larger angle of peak (010) compared to α-cellulose and MCC. The smaller angle of peak (110) for α-cellulose and MCC indicates a lower cellulose Iα content [24, 33]. Furthermore, the differences between Bragg angle of peak at (100) plane and peak at (010) for BC samples show that BC-HP has lower value than BC-FD. This proposes that BC-FD has higher preferences in cellulose Iα compared to BC-HP, thus confirming the previous result obtained from FTIR analysis [33, 34]. The peak deconvolution method for obtaining the crystallinity values for each sample is shown in Fig. 4. From the results, the crystallinity index and crystallinity for BC-FD and BC-HP show a higher value compared to MCC and α-cellulose. The value of crystallinity index for BC-HP was significantly higher than BC-FD which might suggest that drying method applied has an impact to the change in the crystallinity of BC. Furthermore, the value of crystallinity using peak deconvolution method shows similar trends but with lower value. It can be considered the calculation of the crystallinity index using Segal method only for empirical measurement to allow rapid comparison in cellulose crystallinity [17] as the intensity in XRD spectra is not consistent even using the repeated sample. On the other hand, the peak deconvolution method demonstrates a reliable data for the cellulose crystallinity and the comparison of crystallinity value reported by Park et al. [17] was in good agreement with the NMR method. Nevertheless, although the methods between peak intensity and peak deconvolution presented different in crystallinity values, the order of crystallinity for the given samples shows a consistent comparison by each method.

XRD spectra of cellulose samples after background subtraction and baseline correction for cellulose crystallinity calculation using peak deconvolution method. a BC-HP, b BC-FD, c MCC, and d α-cellulose XRD spectra
Table 3 shows that the interplanar distances (d-spacings) for BC-FD and BC-HP have no significant change with each other. Furthermore, the d-spacings for α-cellulose and MCC are observed to give the similar trending with almost similar values. However, the BC samples show greater values for d1 and d2 compared to α-cellulose and MCC, but the d3 values are not obviously different. Additionally, the value obtained for d1, d2, and d3 for all samples is comparable with previous works by Castro et al.[35] and Wada et al. [36]. Moreover, Wada et al. [20] have proposed a simple method for categorizing native celluloses into an Iα-rich type (algal–bacterial) and an Iβ-dominant type (cotton–ramie) by estimating the monoclinic and triclinic relation (Iα/Iβ) from the discriminant analysis of two equatorial d-spacings (d1 and d2) known as Z-value. As presented in Table 3, BC-FD and BC-HP show the Z-value > 0 which indicate that the BC produced was attributed to algal–bacterial group with Iα -rich type, while α-cellulose and MCC demonstrate the Z-value < 0 which belong to cotton–ramie group and Iβ -dominant type. Furthermore, BC-FD shows a slightly higher value of Z-value compared to BC-HP thus suggested that Iα possessed by BC-FD was greater. On the other hand, the crystallite size of BC-HD and BC-FD generally does not show any significant different but can be summarized in the following order: BC-HP > BC-FD > MCC > α-cellulose. This order indicated that BC BC-FD and BC-HP have larger crystallite size compared to α-cellulose and MCC [37]. Additionally, BC-HP possessed larger crystallite size than BC-FD which contributed to the highest crystallinity value, thus distinguishing the effect of drying method in this study.
Thermal analysis using TGA
Figure 5 shows the thermogravimetric (TG) and differential thermal degradation (DTG) curves of cellulose samples depicted from thermogravimetric analysis (TGA). Two main stages of dynamic weight loss were observed for each cellulose sample. The first stage of sample weight loss was considered as dehydration and volatilization of low molecular weight component bounded to cellulosic structures ranging from 5 to 9% at 25–150 °C. The α-cellulose was estimated to have the highest moisture content attributed to higher amorphous region compared to BC-OD, BC-FD, and MCC which have higher crystallinity region. Meanwhile, the highest percentage of weight loss occurred significantly in the second stage of TG graphs which credited to the depolymerization of β-glucan polymeric chains and oxidation of cellulosic materials to carbonaceous residue. From Table 4, the weight loss occurred at the second stage indicates a lower percentage for BC-HP and BC-FD compared to α-cellulose and MCC. Furthermore, the final residue weight percentage for BC-HP and BC-FD was significantly higher than α-cellulose and MCC. These results showed that higher crystallinity content in BC, especially in BC-HP, performs as a flame retardant, thus delaying the thermal decomposition process [38]. Conversely, the facts that BC-FD exhibited higher weight loss and lower residue than BC-HP suggested that the freeze-drying application generated highly porous structure, thus lowering the effect of flame retardant [38, 39]. However, the initial and maximum thermal degradation temperature presented in DTG analysis for BC-HP and BC-FD was considerably lower compared to α-cellulose and MCC. This corresponded to the fraction of cellulose Iα (triclinic) structure in BC-HP and BC-FD that has lower stability than the cellulose Iβ (monoclinic) structure, hence reducing the energy needed thermal decomposition [25]. Thus, it is noteworthy to claim that the BC has higher thermal resistibility compared to others in terms of lower percentage of weight loss and higher in residue.

TGA and DTG thermograph of bacterial cellulose and plant cellulose samples
Thermal analysis through DSC
Further thermal analysis has been carried out using DSC thermograph to elucidate the thermal behavior of cellulose samples as shown in Fig. 6. The earlier peaks occurred at 25–160 °C were representing the endothermic reaction of sample dehydration. In this case, BC-HP shows the lowest dehydration reaction rate followed by BC-FD and MCC with respect to the order of higher crystallinity structures. Meanwhile, the α-cellulose demonstrates the highest dehydration reaction rate compared to others due to physical sponge-like appearances and possessed highest amorphous region that favorably entraps more water molecules in the structures [40]. The next calorimetric peaks that occurred at 270–370 °C indicate melting reaction of crystalline structures and denaturation of glucose units. Surprisingly, BC-HP demonstrated an exothermic reaction peak with the lowest melting point temperature and enthalpy value. It is suspected that the drying approach for the BC-HP resulted to the thin-film appearances similar to the plastic polymer material, thus affecting the thermal melting behavior of the sample [10], whereas BC-FD was representing the endothermic of melting peak comparable to the α-cellulose and MCC due to the physically fibrous porous structures of the samples [11]. Additionally, despite possessing the highest crystallinity values, BC-HP and BC-FD show a lower enthalpy of melting compared to MCC. As mentioned before, BC-HP and BC-FD contain cellulose Iα (triclinic) fractions which has less stability compared to the cellulose Iβ (monoclinic) structure that are dominant in MCC [25]. Overall, BC-HP has lower thermal stability compared to BC-FD and α-cellulose and MCC based on the enthalpy value and melting peak temperature range.

DSC thermograph of cellulose samples. a Overall DSC graph, b peak for dehydration temperature, c peak for melting point temperature
Conclusion
The effect of drying method applied to the BC pellicles on the crystalline structure characteristic and thermal properties was confirmed. FTIR analysis revealed that the fraction of cellulose Iα crystalline structure decreased in BC-HP compared to BC-FD. Meanwhile, the FTIR and XRD analysis on the crystallinity favorably fall into BC-HP compared to BC-FD. Furthermore, the thermal analysis using TGA and DSC demonstrated that BC-HP possessed higher thermal resistivity but lower thermal stability compared to BC-FD. Therefore, applying different methods of drying to the BC pellicles was significantly affected the crystallinity and thermal behavior of BC. Based on this fact, further material application using different approach of drying the BC should particularly take into consideration.
References
Revin V, Liyaskina E, Nazarkina M et al (2018) Cost-effective production of bacterial cellulose using acidic food industry by-products. Brazilian J Microbiol 49:151–159. https://doi.org/10.1016/j.bjm.2017.12.012
Mohammadkazemi F, Azin M, Ashori A (2015) Production of bacterial cellulose using different carbon sources and culture media. Carbohydr Polym 117:518–523. https://doi.org/10.1016/j.carbpol.2014.10.008
Shezad O, Khan S, Khan T, Park JK (2010) Physicochemical and mechanical characterization of bacterial cellulose produced with an excellent productivity in static conditions using a simple fed-batch cultivation strategy. Carbohydr Polym 82:173–180. https://doi.org/10.1016/j.carbpol.2010.04.052
Klemm D, Heublein B, Fink HP, Bohn A (2005) Cellulose: fascinating biopolymer and sustainable raw material. Angew Chemie Int Ed 44:3358–3393. https://doi.org/10.1002/anie.200460587
Lin D, Liu Z, Shen R et al (2020) Bacterial cellulose in food industry: current research and future prospects. Int J Biol Macromol 158:1007–1019. https://doi.org/10.1016/j.ijbiomac.2020.04.230
De AI, Fernandes A, Cristina A et al (2020) Bacterial cellulose: from production optimization to new applications. Int J Biol Macromol 164:2598–2611. https://doi.org/10.1016/j.ijbiomac.2020.07.255
Kosseva MR, Zhong S, Li M et al (2020) Biopolymers produced from food wastes: a case study on biosynthesis of bacterial cellulose from fruit juices. Food Ind Wastes. https://doi.org/10.1016/b978-0-12-817121-9.00011-5
Illa MP, Sharma CS, Khandelwal M (2019) Tuning the physiochemical properties of bacterial cellulose: effect of drying conditions. J Mater Sci 54:12024–12035. https://doi.org/10.1007/s10853-019-03737-9
Clasen C, Sultanova B, Wilhelms T et al (2006) Effects of different drying processes on the material properties of bacterial cellulose membranes. Macromol Symp 244:48–58. https://doi.org/10.1002/masy.200651204
Klemm D, Cranston ED, Fischer D et al (2018) Nanocellulose as a natural source for groundbreaking applications in materials science: today’s state. Mater Today 21:720–748. https://doi.org/10.1016/j.mattod.2018.02.001
Andree V, Niopek D, Müller C et al (2021) Influence of drying methods on the physical properties of bacterial nanocellulose. Mater Res Express. https://doi.org/10.1088/2053-1591/abe016
Hashim NA, Zakaria J, Mohamad S, Mohamad SF et al (2021) Effect of different treatment methods on the purification of bacterial cellulose produced from OPF juice by Acetobacter Xylinum. IOP Conf Ser Mater Sci Eng 1092:012058. https://doi.org/10.1088/1757-899x/1092/1/012058
Sharifah Fathiyah SM, Shahril M, Junaidi Z (2021) Oil palm frond juice and coconut water as alternative fermentation substrate for bacterial cellulose production. IOP Conf Ser Mater Sci Eng 1092:012055. https://doi.org/10.1088/1757-899x/1092/1/012055
Nada AMA, Shabaka AA, Yousef MA, Abd-El-Nour KN (1990) Infrared spectroscopic and dielectric studies of swollen cellulose. J Appl Polym Sci 40:731–739. https://doi.org/10.1002/app.1990.070400510
Kataoka Y, Kondo T (1999) Quantitative analysis for the cellulose Iα crystalline phase in developing wood cell walls. Int J Biol Macromol 24:37–41. https://doi.org/10.1016/S0141-8130(98)00065-8
Segal L, Creely JJ, Martin AE, Conrad CM (1959) An Empirical method for estimating the degree of crystallinity of native cellulose using the X-Ray diffractometer. Text Res J 29:786–794. https://doi.org/10.1177/004051755902901003
Park S, Baker JO, Himmel ME et al (2010) Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3:1–10. https://doi.org/10.1186/1754-6834-3-10
Bragg WH, Bragg WL (1913) The reflection of X-rays by crystals. Proc R Soc Lond A 88:428–438. https://doi.org/10.1098/rspa.1913.0040
Patterson AL (1939) The scherrer formula for X-ray particle size determination. Phys Rev 56:978–982. https://doi.org/10.1103/PhysRev.56.978
Wada M, Okano T, Sugiyama J (2001) Allomorphs of native crystalline cellulose I evaluated by two equatorial d-spacings. J Wood Sci 47:124–128. https://doi.org/10.1007/BF00780560
Revin VV, Pestov NA, Shchankin MV et al (2019) A study of the physical and mechanical properties of aerogels obtained from bacterial cellulose. Biomacromol 20:1401–1411. https://doi.org/10.1021/acs.biomac.8b01816
O’connor RT, Dupré EF, Mitcham D (1958) Applications of infrared absorption spectroscopy to investigations of cotton and modified cottons: part I—physical and crystalline modifications and oxidation. Text Res J 28:382–392. https://doi.org/10.1177/004051755802800503
Andritsou V, De Melo EM, Tsouko E et al (2018) Synthesis and characterization of bacterial cellulose from citrus-based sustainable resources. ACS Omega 3:10365–10373. https://doi.org/10.1021/acsomega.8b01315
Sugiyama J, Vuong R, Chanzy H (1991) Electron diffraction study on the two crystalline phases occurring in native cellulose from an algal cell wall. Macromolecules 24:4168–4175. https://doi.org/10.1021/ma00014a033
Heiner AP, Sugiyama J, Teleman O (1995) Crystalline cellulose Iα and Iβ studied by molecular dynamics simulation. Carbohydr Res 273:207–223. https://doi.org/10.1016/0008-6215(95)00103-Z
Vazquez A, Foresti ML, Cerrutti P, Galvagno M (2013) Bacterial cellulose from simple and low cost production media by Gluconacetobacter xylinus. J Polym Environ 21:545–554. https://doi.org/10.1007/s10924-012-0541-3
Qiao N, Fan X, Zhang X et al (2019) Soybean oil refinery effluent treatment and its utilization for bacterial cellulose production by Gluconacetobacter xylinus. Food Hydrocoll. https://doi.org/10.1016/j.foodhyd.2019.105185
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896. https://doi.org/10.1007/s10570-013-0030-4
Chen G, Wu G, Chen L et al (2019) Performance of nanocellulose-producing bacterial strains in static and agitated cultures with different starting pH. Carbohydr Polym 215:280–288. https://doi.org/10.1016/j.carbpol.2019.03.080
Agarwal UP, Ralph SA, Baez C et al (2017) Effect of sample moisture content on XRD-estimated cellulose crystallinity index and crystallite size. Cellulose 24:1971–1984. https://doi.org/10.1007/s10570-017-1259-0
Andersson S, Serimaa R, Paakkari T et al (2003) Crystallinity of wood and the size of cellulose crystallites in Norway spruce (Picea abies). J Wood Sci 49:531–537. https://doi.org/10.1007/s10086-003-0518-x
Tyagi N, Suresh S (2016) Production of cellulose from sugarcane molasses using Gluconacetobacter intermedius SNT-1: optimization & characterization. J Clean Prod 112:71–80. https://doi.org/10.1016/j.jclepro.2015.07.054
Watanabe K, Tabuchi M, Morinaga Y, Yoshinaga F (1998) Structural features and properties of bacterial cellulose produced in agitated culture. Cellulose 5:187–200. https://doi.org/10.1023/A:1009272904582
Chen QJ, Zhou LL, Zou JQ, Gao X (2019) The preparation and characterization of nanocomposite film reinforced by modified cellulose nanocrystals. Int J Biol Macromol 132:1155–1162. https://doi.org/10.1016/j.ijbiomac.2019.04.063
Castro C, Zuluaga R, Putaux JL et al (2011) Structural characterization of bacterial cellulose produced by Gluconacetobacter swingsii sp. from Colombian agroindustrial wastes. Carbohydr Polym 84:96–102. https://doi.org/10.1016/j.carbpol.2010.10.072
Wada M, Okano T (2001) Localization of Iα and Iβ phases in algal cellulose revealed by acid treatments. Cellulose 8:183–188. https://doi.org/10.1023/A:1013196220602
Wada M, Okano T, Sugiyama J (1997) Synchrotron-radiated X-ray and neutron diffraction study of native cellulose. Cellulose 4:221–232. https://doi.org/10.1023/A:1018435806488
Dai H, Ou S, Huang Y, Huang H (2018) Utilization of pineapple peel for production of nanocellulose and film application. Cellulose 25:1743–1756. https://doi.org/10.1007/s10570-018-1671-0
Blanco A, Monte MC, Campano C et al (2018) Nanocellulose for industrial use. Elsevier
Shah N, Ul-Islam M, Khattak WA, Park JK (2013) Overview of bacterial cellulose composites: a multipurpose advanced material. Carbohydr Polym 98:1585–1598. https://doi.org/10.1016/j.carbpol.2013.08.018
Acknowledgements
The research was funded by the Ministry of Higher Education under Fundamental Research Grant Scheme No. FRGS/1/2018/TK05/UMP/03/3 (RDU190154). The authors would like to acknowledge Universiti Putra Malaysia (UPM) and Universiti Malaysia Pahang (UMP) for the technical support during the study period. Finally, our gratitude also goes to UMP and MOHE for the provision of study leave and scholarship for the first author.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Additional information
Handling Editor: Stephen Eichhorn.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mohamad, S., Abdullah, L.C., Jamari, S.S. et al. Influence of drying method on the crystal structure and thermal property of oil palm frond juice-based bacterial cellulose. J Mater Sci 57, 1462–1473 (2022). https://doi.org/10.1007/s10853-021-06685-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-021-06685-5