
Abstract
New evidence has emerged in recent years to suggest a strong link between the human gut microbiota, its metabolites, and various physiological aspects of hosts along with important pathophysiological dimensions of diseases. The research indicates that the gut microbiota can facilitate metabolite production in two ways: first, the resident species of the gut microbiota use the amino acids produced from food or the host as elements for protein synthesis, and second, conversion or fermentation are used to drive nutrient metabolism. Additionally, the gut microbiota can synthesize several nutritionally essential amino acids de novo, which is a potential regulatory factor in amino acid homeostasis. The primary objective of this review is to summarize the current literature relating to the ways in which microbial amino acids contribute to host amino acid homeostasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Host homeostasis with respect to issues of physiology and metabolism is crucially underpinned by the gut microbiota and its metabolites (Human Microbiome Project C 2012). Recent literature indicates that various quantitative and qualitative transformations associated with several metabolites generated by the gut microbiota are implicated in the pathophysiological dimensions of illnesses such as metabolic syndrome, inflammatory bowel disease, diabetes mellitus (types 1 and 2), asthma, colon cancer, obesity, major depression, and autism (Diaz Heijtz et al. 2011; Vijay-Kumar et al. 2010; Uronis et al. 2009; De Filippo et al. 2010).
A fundamental function of the gut microbiota that has long been understood by the research community is the metabolism of indigestible matter consumed by the host, thereby contributing to optimal energy production. In this context, as a prominent part of the human diet, amino acids play a crucial role not simply by serving as the basic elements of proteins and peptides, but more importantly in driving the production of numerous bioactive molecules that contribute to the maintenance of signaling pathways and metabolism (Sato et al. 2006; Wu 2013; Wu et al. 2014). Researchers have compared germ-free mice and conventionalized mice and found that the latter possessed an altered distribution of free amino acids in the gastrointestinal (GI) tract, importantly indicating that the resident species of the gut microbiota are crucial to host amino acid homeostasis and health (Mardinoglu et al. 2015). The purpose of the present review is to outline how host nutritional status and physiological health are prominently influenced by amino acid metabolism in the gut microbiota.
Microbial amino acid metabolism
Gut microbiota and the regulation of amino acid catabolism and utilization
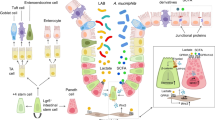
The gut microbiota performs a crucial function in facilitating the regulation of the amino acid pool and profile over the course of amino acid digestion and absorption. It should be noted that the amino acid pool refers to the overall number of amino acids, whereas the amino acid profile refers to the compositional features of amino acids on an individual basis. It is also noteworthy that a comparative examination of germ-free and conventionalized mice demonstrated that the resident bacterial species of the gut influenced the free amino acid distribution in the GI tract (Mardinoglu et al. 2015).
Using the standard microbiological technique of plate counting, studies in the literature have also reported that the bacterial species that predominate in using single amino acids or pairs of amino acids are effectively distinct. Research showed that in milk-fed piglets the microbiota of the small intestine draws on lysine (Han et al. 2017). Furthermore, the researchers determined that lysine catabolism inside the intestinal mucosa occurs at a higher rate than lysine absorption through the mucosa. Evenepoel et al. (1999) determined the efficiency of protein breakdown prior to amino acid absorption in the small intestine, and noted that significant quantities of amino acids are not assimilated in humans. In recently conducted research addressing the resident bacterial species within the human colon, the findings indicated substantial quantities of protein- and amino acid-fermenting bacteria. Specifically, bacteria of the Clostridium genus located in the large intestine (the fundamental bacteria for lysine or proline utilization) are the key driver of amino acid fermentation, whereas bacteria of the Peptostreptococcus genus are the key driver of glutamate or tryptophan use. Nevertheless, it is important to note that several species could play a prominent role in amino acid metabolism in the large intestine, such as bacteria of the genera Fusobacterium, Bacteroides, and Veillonella and the species Megasphaera elsdenii and Selenomonas ruminantium (Dai et al. 2011).
In view of this, the fundamental consideration is that amino acid utilization can be directed toward the production of bacterial cell components and, furthermore, that amino acids can be catabolized through distinct pathways. Moreover, the diverse nature of amino acid metabolism in the resident species of the gut microbiota can result in beneficial or adverse impacts on the host.
The impact of dietary protein on the gut microbiota
The overall profile of the gut microbiota is influenced by numerous variables, including the following: (1) host-based factors (genetics, digestive secretions, digestive physiology, diet, health, drug use, and innate and adaptive immunity); (2) microbiological factors (nutrient and adhesion site competition, metabolic cooperation, and bacterial antagonism); and (3) environmental factors (local pH, the presence of substrates, redox potential, and geography). One of the fundamental variables in this respect is the host diet, which has been shown to have a profound impact on the profile and operation of the microbiota of the GI tract (Rist et al. 2013). Moderating dietary proteins or amino acids could represent a strategic approach for the control of amino acid-fermenting bacterial species and their metabolic pathways, which in turn could have an impact on the metabolism of the host. In view of this, it is important to consider the previous research projects that have sought to determine the effects of dietary protein sources on the overall profile of the gut microbiota.
Shen et al. (2010) examined human fecal batch cultures and revealed a significant rise in Clostridium perfringens combined with a significant fall in bacteria of the Bifidobacteria genus after 2 days of incubation with beef protein. Milk-fed piglets that after weaning were sustained on a diet of dried skimmed milk powder consumed a greater amount of feed than their counterparts fed a diet of soybean meal (SBM), thereby resulting in a corresponding increase in body weight (BW) and a favorable feed conversion ratio (Rist et al. 2013). Despite the good health status of the piglets in both the milk-fed and SBM diet categories, those in the latter group, owing to the buffering effect of the protein, exhibited a higher gastrointestinal pH and a lower lactobacillus-to-coliform ratio (Partanen and Mroz 1999). A higher lactobacillus-to-coliform ratio indicates a favorable proportion of advantageous lactobacilli relative to coliforms, which can incorporate coliform pathogens (Rist et al. 2013). It is also notable that interventional research projects have been conducted to comparatively examine the bacterial profile modifications in certain sample groups depending on dietary protein content (Vital et al. 2014). For example, a sample of individuals in full health was examined over a period of 42 days with one group consuming a high-protein diet and the other consuming a low-protein diet. The results revealed no change in the proportions of bacteria in the Clostridium and Bacteroides genera, or sulfate-reducing bacterial species, although a reduction in the pool of bacteria belonging to the Bifidobacteria genus and others was observed (Brinkworth et al. 2009; Duncan et al. 2007). It is clear that dietary factors (specifically, protein consumption) can enhance gastrointestinal health by shaping the nature of the gut microbiota (Fig. 1).

The fate of protein catabolism associated with the gut microbiota
The biosynthesis of microbial amino acids
Amino acids are the fundamental elements of proteins and peptides and are crucial for the production of numerous bioactive molecules that play regulatory roles in signaling pathways and metabolism (Dai et al. 2015). In addition to the utilization of amino acids, the gut microbiota performs a key function in producing amino acids, and this includes de novo biosynthesis. For instance, several in vitro research projects have indicated that ruminal bacterial species, such as Streptococcus bovis, Selenomonas ruminantium, and Prevotella bryantii, engage in the de novo synthesis of amino acids in the presence of physiological peptide concentrations (Hullar and Fu 2014). In addition, other in vivo research projects have demonstrated that microbially derived lysine is absorbed and integrated into the host proteins (Ciarlo et al. 2016; Hullar and Fu 2014). Consistent with these animal-based research projects, the oral consumption of 15N in the form of 15NH4Cl by a sample of six males in full health was associated with the labeling of microbial proteins and threonine from intestinal microbial origin, which was present in the portal bloodstream in vivo (Metges et al. 1999). In addition, Metges et al. (1999) examined a sample of humans over the age of 18 who were supplied with nitrogen-adequate diets and revealed that microbially derived lysine and threonine contribute significantly to the free plasma lysine and threonine pool. Moreover, Gill et al. (2006) reported that the enrichment of the microbiota in the large intestine takes place via genes implicated in essential amino acid (EAA) biosynthesis, which occurs based on precursors generated from the human plasma pool.
Amino acids as precursors for microbially derived short-chain fatty acids
It has been demonstrated in the literature that short-chain fatty acids (SCFAs) constitute the fundamental output of fermentation (Hijova and Chmelarova 2007; Wong et al. 2006). In this context, it is notable that undigested proteins and amino acids within the colon have the potential to operate as a supplemental substrate for SCFA generation, in addition to indigestible carbohydrates (Ciarlo et al. 2016). In terms of the roles played by microorganisms within the colon and, moreover, the biological mechanisms that they moderate, a central and crucial physiological process is SCFA synthesis, where several amino acids generated from microbial protein fermentation in the large intestine function as synthetic precursors to SCFAs (Ciarlo et al. 2016; Mu et al. 2017). Numerous amino acids employed by anaerobic bacteria have the potential to be metabolized to acetate, including glycine, threonine, glutamate, and ornithine (Neis et al. 2015), whereas threonine, lysine, and glutamate can be utilized for butyrate synthesis. Davila et al. (2013) reported that propionate is primarily synthesized from threonine. These results indicate that of all of the amino acids utilized for SCFA synthesis, threonine—as it produces each of the three fundamental SCFAs—possesses the greatest adaptability.
As the literature indicates, SCFAs—the most abundant of which are acetate, propionate, and butyrate—play numerous biological roles: (1) they constitute an energy source for muscles, the kidneys, the cardiovascular system, and the brain; (2) they facilitate hepatic control of lipids and carbohydrates; (3) they are implicated in the transportation and metabolism of epithelial cells; and (4) they influence epithelial cell growth and differentiation (Macfarlane and Macfarlane 2012). The other SCFAs that are generated by resident species in the gut microbiota include valerate, isovalerate, 2-methyl butyrate, and formate, but it is important to note that their production is comparatively lower.
The gut microbiota and amino acid perturbations in the progression of obesity and type 2 diabetes mellitus
Evidence is beginning to mount to support the hypothesis that changes to the profile and operation of the gut microbiota can lead to the onset and progression of various diseases, including insulin resistance, obesity, and type 2 diabetes mellitus (henceforth, type 2 DM). For sufferers of type 2 DM, it is important to recognize that a modified Firmicutes-to-Bacteroides ratio is correlated with higher levels of energy harvesting and, subsequently, the onset of insulin resistance (Tilg and Kaser 2011; Tremaroli and Backhed 2012). Obesity affects the profile of the gut microbiota (Turnbaugh et al. 2009), with one of the key differences being greater and lesser proportions of Firmicutes and Bacteroidetes, respectively (Ley et al. 2006; Ley 2010). Taken together, these results indicate that certain bacterial genera are related to an individual’s BMI, which in turn suggests that gut dysbiosis is connected to the progression of disease, in particular, type 2 DM. Researchers have reported a strong correlation between type 2 DM and increased and decreased proportions of Clostridium clostridioforme and Roseburia 272, respectively, in Chinese and European sample groups (Karlsson et al. 2013; Qin et al. 2012).
In view of these findings, it is clear that more in-depth investigations should be performed into the possible roles of the gut microbiota in amino acid and SCFA perturbations. Nevertheless, despite the need for more data regarding the physiological processes involved, relatively concrete conclusions can already be drawn. The functional products of the gut microbiota (especially bacterial metabolites such as SCFAs and amino acids) play a critical role in mediating the physiological aspects of the host (Sridharan et al. 2014). It is possible to conclude that dysbiosis, which contributes to a downscaling of SCFA synthesis, can result in obesity and the translocation of lipopolysaccharides into the systemic circulation, which probably explains the appearance of metabolic endotoxemia in metabolic syndrome patients, in addition to those with type 2 DM and insulin resistance. Research focused on humans over the age of 18 suggests that obesity, insulin resistance, and type 2 DM are associated with high systemic concentrations of branched-chain amino acids (BCAAs) (Wang et al. 2011). Amino acid changes of this kind, according to the available evidence, are implicated in metabolic disorders; for example, lower insulin sensitivity was induced in one male patient by applying 18 amino acids (such as the BCAAs leucine, isoleucine, and valine) (Tremblay et al. 2005). Additionally, Do et al. (2014) revealed that a significant rise in the portal concentrations of a range of EAAs occurs for subjects with obesity and glucose intolerance stemming from a high-fat diet.
It is also notable that elevated Roseburia levels after fecal transplants from healthy weight individuals to those with metabolic syndrome appear to have contributed to favorable insulin sensitivity (Vrieze et al. 2012). Other prominent results include those from clinical research projects in which the direct supplementation of advantageous microbes was found to have the potential to mediate host hepatic and systemic lipid metabolism (Christ et al. 2015), energy homeostasis (Backhed et al. 2007), and glycemic control (Mazloom et al. 2013). Each of these advantages is also associated with fecal microbial transplants (Vrieze et al. 2012), and they are associated with decreased risks of diet-based obesity, insulin resistance, and type 2 DM.
Microbially derived tryptophan catabolites in central nervous system function
Research has indicated that the profile of the gut microbiota is the key determinant of the levels of tryptophan catabolites (TRYCATs) in the systemic circulation. Consequently, the profile of the gut microbiota is an indirect contributor to the degree to which serotonin is present in the brain. In the course of illnesses linked to microbiota dysbiosis (e.g., ileitis), the utilization of tryptophan in the GI tract is affected, resulting in elevated luminal tryptophan levels and a concurrent decrease in tryptophan metabolites (Hisamatsu et al. 2012; Schicho et al. 2010). Consequently, the dominant capacity of the microbiota to facilitate the regulation of tryptophan metabolism throughout the TRYCAT pathway results in the following: (1) the activity reduction of pathways implicated in serotonin synthesis; and (2) an increase in the production of quinolinic acid and kynurenic acid, along with various neuroactive metabolites (Stone et al. 2013; Mawe and Hoffman 2013). As serotonin is a fundamental neurotransmitter at every signaling terminus of the gut–brain axis (O’Mahony et al. 2015), this is a consequential fact. Moreover, in addition to serving as a building block for proteins, tryptophan, as an amino acid, is a substrate for host-based metabolic biotransformation into numerous chemoeffectors (e.g., the neurotransmitters serotonin and melatonin, trace quantities of niacin, and indoleamine-2,3-dioxygenase [IDO)-dependent kynurenines (AhR ligands)] (Nguyen et al. 2014). Aberrances regarding TRYCATs are known to play a role in the onset and progression of depression, chronic fatigue syndrome, and somatoform disorder (Maes et al. 2007, 2011; Maes and Rief 2012), and aberrances in the levels of neurotoxic and immunomodulatory TRYCATs have been observed in several neurodegenerative and neuroimmune diseases (Morris et al. 2016). In addition to the host-based metabolism of tryptophan, the gut microbiota facilitates distinctive catabolic biotransformations of tryptophan that are involved in the production of a series of bioactive metabolites. In addition, the microbiota has been implicated in influencing the levels of γ-aminobutyric acid (GABA), brain-derived neurotrophic factor (BDNF), and noradrenaline and dopamine (Diaz Heijtz et al. 2011; Keightley et al. 2015). It should be noted that these are important metabolites with respect to the interface between the GI tract and the brain. Ultimately, the existence of neuroactive metabolites of this kind in the systemic circulation has explanatory power in accounting for the importance of the microbiota in the operation and evolution of the central nervous system. Furthermore, it can help to elucidate the various ways in which the microbiota plays a role in affecting brain function in such a way as to produce neurological disorders.
Amino acid-metabolizing bacteria and pregnancy
The capacity of the GI tract to adapt over the course of pregnancy is crucial in allowing the female to accommodate the evolving requirements of the fetus. Researchers have found that for mammalian species, the profile of the gut microbiota transforms during pregnancy in line with the overall modification of the body’s metabolic status (Koren et al. 2012; Collado et al. 2008; Santacruz et al. 2010). In human females, over the course of the first trimester, the profile of the gut microbiota is comparable to that observed in a healthy female who is not pregnant. However, by the third trimester, the key characteristics of the gut microbiota include elevated levels of Proteobacteria and Actinobacteria (in particular, bacteria of the Streptococcus and Enterobacteriaceae genera) (Koren et al. 2012). Other changes during late pregnancy include a reduction in the quantity of bacteria of the Faecalibacterium genus (Haro et al. 2016). Following birth (specifically, during preliminary lactation/postpartum), the level of bacteria of the Streptococcus genus usually falls, although it remains elevated in relation to the first trimester (Koren et al. 2012; Jost et al. 2014). Hence, it is possible to conclude that the GI microbiome of a human female is subject to significant modification over the course of pregnancy, and the overall reduction in microbial diversity is comparable to that observed during obesity (Qin et al. 2010; Greenblum et al. 2012). The defining characteristic of the microbiome of an obese individual is a higher capacity for energy utilization, which is mainly due to the increased level of bacteria that can drive fermentation. Ultimately, this increases the degree to which usually indigestible sugars are available.
In view of these observations, it is evident that the wealth of bacterial species associated with intestinal amino acid metabolism is subject to a further increase in late gestation and, furthermore, in pregnant women who weigh more than they should. Several explanations for this have been suggested in the literature: (1) the control of protein and amino acid digestion and absorption in the small intestine; (2) the degree to which dietary amino acids are available for utilization by the reproductive organs; (3) long-term modifications in the profile and total bacterial count of the microbiota over the course of pregnancy; (4) modifications to uterine capacity and operation, along with the metabolic nature of the body; (5) the implementation of metabolic transformations in the gut microbiota of both the female and the child; and (6) modifications in the synthesis of amino acid metabolites implicated in reproductive processes (e.g., NO and glutathione). Note that this last effect takes place in a direct or indirect way in line with the physiological aspects of the host’s reproductive system. Nevertheless, it should not be overlooked that the underlying mechanisms through which such modifications occur are not yet understood, emphasizing the necessity of further research in this field.
Conclusion
As evidence regarding the impact of the human gut microbiota on the health of the gastrointestinal tract continues to grow, the significant correlation between these factors is becoming indisputable (Klose et al. 2010). Moreover, it is becoming clear that a highly responsive and mutual connection exists between the resident species of the gut microbiota and the host, primarily insofar as the host metabolome status is regulated by the microbiota locally and systemically (Zhang et al. 2015). In view of this, the present review summarizes the existing literature relating to the ways in which microbial amino acids contribute to host amino acid homeostasis.
In this active field of research, numerous findings have been reported in recent years regarding the impact that resident bacterial species in the gut have on the metabolism and recycling of dietary amino acids. It has become clear that these bacteria influence the entry of amino acids into the portal circulation for whole-body use (Hullar and Fu 2014). In addition, it has been demonstrated that bacteria colonizing the intestine have the capacity to facilitate the de novo synthesis of EAAs, which are implicated in amino acid homeostasis in the host (Collins et al. 2012).
Disruptions to the human gut microbiota have widespread implications. For instance, they are associated with diseases external to the intestine, including the systemic immune system, the genital system, the central nervous system, and adipose tissues (Ciarlo et al. 2016). In view of this, it is clear that more complete knowledge of microbiota metabolite diversity and operation will be crucial for developing new treatment interventions for relevant diseases. Nevertheless, the difficulty in discerning microbiota metabolites, and the complex nature of the procedures and processes through which the physiological aspects of a host are regulated by the microbiota are fundamental obstacles. Future research must concomitantly examine the metabolome and microbiome relating to health and illness to completely illuminate the connection between microbiota composition and function.
References
Backhed F, Manchester JK, Semenkovich CF, Gordon JI (2007) Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA 104(3):979–984. doi:10.1073/pnas.0605374104
Brinkworth GD, Noakes M, Clifton PM, Bird AR (2009) Comparative effects of very low-carbohydrate, high-fat and high-carbohydrate, low-fat weight-loss diets on bowel habit and faecal short-chain fatty acids and bacterial populations. Br J Nutr 101(10):1493–1502. doi:10.1017/S0007114508094658
Christ B, Bruckner S, Winkler S (2015) The therapeutic promise of mesenchymal stem cells for liver restoration. Trends Mol Med 21(11):673–686. doi:10.1016/j.molmed.2015.09.004
Ciarlo E, Heinonen T, Herderschee J, Fenwick C, Mombelli M, Le Roy D, Roger T (2016) Impact of the microbial derived short chain fatty acid propionate on host susceptibility to bacterial and fungal infections in vivo. Sci Rep UK. doi:10.1038/srep37944
Collado MC, Isolauri E, Laitinen K, Salminen S (2008) Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr 88(4):894–899
Collins SM, Surette M, Bercik P (2012) The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol 10(11):735–742. doi:10.1038/nrmicro2876
Dai ZL, Wu G, Zhu WY (2011) Amino acid metabolism in intestinal bacteria: links between gut ecology and host health. Front Biosci (Landmark Ed) 16:1768–1786
Dai Z, Wu Z, Hang S, Zhu W, Wu G (2015) Amino acid metabolism in intestinal bacteria and its potential implications for mammalian reproduction. Mol Hum Reprod 21(5):389–409. doi:10.1093/molehr/gav003
Davila AM, Blachier F, Gotteland M, Andriamihaja M, Benetti PH, Sanz Y, Tome D (2013) Re-print of “Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host”. Pharmacol Res 69(1):114–126. doi:10.1016/j.phrs.2013.01.003
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA 107(33):14691–14696. doi:10.1073/pnas.1005963107
Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S (2011) Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA 108(7):3047–3052. doi:10.1073/pnas.1010529108
Do TT, Hindlet P, Waligora-Dupriet AJ, Kapel N, Neveux N, Mignon V, Delomenie C, Farinotti R, Feve B, Buyse M (2014) Disturbed intestinal nitrogen homeostasis in a mouse model of high-fat diet-induced obesity and glucose intolerance. Am J Physiol Endocrinol Metab 306(6):E668–E680. doi:10.1152/ajpendo.00437.2013
Duncan SH, Belenguer A, Holtrop G, Johnstone AM, Flint HJ, Lobley GE (2007) Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl Environ Microbiol 73(4):1073–1078. doi:10.1128/AEM.02340-06
Evenepoel P, Claus D, Geypens B, Hiele M, Geboes K, Rutgeerts P, Ghoos Y (1999) Amount and fate of egg protein escaping assimilation in the small intestine of humans. Am J Physiol 277(5 Pt 1):G935–G943
Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE (2006) Metagenomic analysis of the human distal gut microbiome. Science 312(5778):1355–1359. doi:10.1126/science.1124234
Greenblum S, Turnbaugh PJ, Borenstein E (2012) Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci USA 109(2):594–599. doi:10.1073/pnas.1116053109
Han GG, Lee JY, Jin GD, Park J, Choi YH, Chae BJ, Kim EB, Choi YJ (2017) Evaluating the association between body weight and the intestinal microbiota of weaned piglets via 16S rRNA sequencing. Appl Microbiol Biotechnol 101(14):5903–5911. doi:10.1007/s00253-017-8304-7
Haro C, Garcia-Carpintero S, Alcala-Diaz JF, Gomez-Delgado F, Delgado-Lista J, Perez-Martinez P, Rangel Zuniga OA, Quintana-Navarro GM, Landa BB, Clemente JC, Lopez-Miranda J, Camargo A, Perez-Jimenez F (2016) The gut microbial community in metabolic syndrome patients is modified by diet. J Nutr Biochem 27:27–31. doi:10.1016/j.jnutbio.2015.08.011
Hijova E, Chmelarova A (2007) Short chain fatty acids and colonic health. Bratisl Lek Listy 108(8):354–358
Hisamatsu T, Okamoto S, Hashimoto M, Muramatsu T, Andou A, Uo M, Kitazume MT, Matsuoka K, Yajima T, Inoue N, Kanai T, Ogata H, Iwao Y, Yamakado M, Sakai R, Ono N, Ando T, Suzuki M, Hibi T (2012) Novel, objective, multivariate biomarkers composed of plasma amino acid profiles for the diagnosis and assessment of inflammatory bowel disease. PLoS One 7(1):e31131. doi:10.1371/journal.pone.0031131
Hullar MAJ, Fu BC (2014) Diet, the gut microbiome, and epigenetics. Cancer J 20(3):170–175
Human Microbiome Project C (2012) Structure, function and diversity of the healthy human microbiome. Nature 486(7402):207–214. doi:10.1038/nature11234
Jost T, Lacroix C, Braegger C, Chassard C (2014) Stability of the maternal gut microbiota during late pregnancy and early lactation. Curr Microbiol 68(4):419–427. doi:10.1007/s00284-013-0491-6
Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, Nielsen J, Backhed F (2013) Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498(7452):99–103. doi:10.1038/nature12198
Keightley PC, Koloski NA, Talley NJ (2015) Pathways in gut-brain communication: evidence for distinct gut-to-brain and brain-to-gut syndromes. Aust NZ J Psychiatry 49(3):207–214. doi:10.1177/0004867415569801
Klose V, Bayer K, Bruckbeck R, Schatzmayr G, Loibner AP (2010) In vitro antagonistic activities of animal intestinal strains against swine-associated pathogens. Vet Microbiol 144(3–4):515–521. doi:10.1016/j.vetmic.2010.02.025
Koren O, Goodrich JK, Cullender TC, Spor A, Laitinen K, Backhed HK, Gonzalez A, Werner JJ, Angenent LT, Knight R, Backhed F, Isolauri E, Salminen S, Ley RE (2012) Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150(3):470–480. doi:10.1016/j.cell.2012.07.008
Ley RE (2010) Obesity and the human microbiome. Curr Opin Gastroenterol 26(1):5–11. doi:10.1097/MOG.0b013e328333d751
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444(7122):1022–1023. doi:10.1038/4441022a
Macfarlane GT, Macfarlane S (2012) Bacteria, colonic fermentation, and gastrointestinal health. J AOAC Int 95(1):50–60
Maes M, Rief W (2012) Diagnostic classifications in depression and somatization should include biomarkers, such as disorders in the tryptophan catabolite (TRYCAT) pathway. Psychiatry Res 196(2–3):243–249. doi:10.1016/j.psychres.2011.09.029
Maes M, Mihaylova I, Ruyter MD, Kubera M, Bosmans E (2007) The immune effects of TRYCATs (tryptophan catabolites along the IDO pathway): relevance for depression—and other conditions characterized by tryptophan depletion induced by inflammation. Neuro Endocrinol Lett 28(6):826–831
Maes M, Leonard BE, Myint AM, Kubera M, Verkerk R (2011) The new ‘5-HT’ hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog Neuropsychopharmacol Biol Psychiatry 35(3):702–721. doi:10.1016/j.pnpbp.2010.12.017
Mardinoglu A, Shoaie S, Bergentall M, Ghaffari P, Zhang C, Larsson E, Backhed F, Nielsen J (2015) The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol Syst Biol 11(10):834. doi:10.15252/msb.20156487
Mawe GM, Hoffman JM (2013) Serotonin signalling in the gut—functions, dysfunctions and therapeutic targets. Nat Rev Gastroenterol Hepatol 10(8):473–486. doi:10.1038/nrgastro.2013.105
Mazloom Z, Yousefinejad A, Dabbaghmanesh MH (2013) Effect of probiotics on lipid profile, glycemic control, insulin action, oxidative stress, and inflammatory markers in patients with type 2 diabetes: a clinical trial. Iran J Med Sci 38(1):38–43
Metges CC, El-Khoury AE, Henneman L, Petzke KJ, Grant I, Bedri S, Pereira PP, Ajami AM, Fuller MF, Young VR (1999) Availability of intestinal microbial lysine for whole body lysine homeostasis in human subjects. Am J Physiol 277(4 Pt 1):E597–E607
Morris G, Berk M, Carvalho A, Caso JR, Sanz Y, Walder K, Maes M (2016) The role of the microbial metabolites including tryptophan catabolites and short chain fatty acids in the pathophysiology of immune-inflammatory and neuroimmune disease. Mol Neurobiol. doi:10.1007/s12035-016-0004-2
Mu CL, Yang YX, Yu KF, Yu M, Zhang CJ, Su Y, Zhu WY (2017) Alteration of metabolomic markers of amino-acid metabolism in piglets with in-feed antibiotics. Amino Acids 49(4):771–781. doi:10.1007/s00726-017-2379-4
Neis EP, Dejong CH, Rensen SS (2015) The role of microbial amino acid metabolism in host metabolism. Nutrients 7(4):2930–2946. doi:10.3390/nu7042930
Nguyen NT, Nakahama T, Le DH, Van Son L, Chu HH, Kishimoto T (2014) Aryl hydrocarbon receptor and kynurenine: recent advances in autoimmune disease research. Front Immunol 5:551. doi:10.3389/fimmu.2014.00551
O’Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF (2015) Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res 277:32–48. doi:10.1016/j.bbr.2014.07.027
Partanen KH, Mroz Z (1999) Organic acids for performance enhancement in pig diets. Nutr Res Rev 12(1):117–145. doi:10.1079/095442299108728884
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Meta HITC, Bork P, Ehrlich SD, Wang J (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464(7285):59–65. doi:10.1038/nature08821
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490(7418):55–60. doi:10.1038/nature11450
Rist VT, Weiss E, Eklund M, Mosenthin R (2013) Impact of dietary protein on microbiota composition and activity in the gastrointestinal tract of piglets in relation to gut health: a review. Anim Int J Anim Biosci 7(7):1067–1078. doi:10.1017/S1751731113000062
Santacruz A, Collado MC, Garcia-Valdes L, Segura MT, Martin-Lagos JA, Anjos T, Marti-Romero M, Lopez RM, Florido J, Campoy C, Sanz Y (2010) Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr 104(1):83–92. doi:10.1017/S0007114510000176
Sato N, Moore FA, Kone BC, Zou L, Smith MA, Childs MA, Moore-Olufemi S, Schultz SG, Kozar RA (2006) Differential induction of PPAR-gamma by luminal glutamine and iNOS by luminal arginine in the rodent postischemic small bowel. Am J Physiol Gastrointest Liver Physiol 290(4):G616–G623. doi:10.1152/ajpgi.00248.2005
Schicho R, Nazyrova A, Shaykhutdinov R, Duggan G, Vogel HJ, Storr M (2010) Quantitative metabolomic profiling of serum and urine in DSS-induced ulcerative colitis of mice by (1)H NMR spectroscopy. J Proteome Res 9(12):6265–6273. doi:10.1021/pr100547y
Shen Q, Chen YA, Tuohy KM (2010) A comparative in vitro investigation into the effects of cooked meats on the human faecal microbiota. Anaerobe 16(6):572–577. doi:10.1016/j.anaerobe.2010.09.007
Sridharan GV, Choi K, Klemashevich C, Wu C, Prabakaran D, Pan LB, Steinmeyer S, Mueller C, Yousofshahi M, Alaniz RC, Lee K, Jayaraman A (2014) Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat Commun 5:5492. doi:10.1038/ncomms6492
Stone TW, Stoy N, Darlington LG (2013) An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol Sci 34(2):136–143. doi:10.1016/j.tips.2012.09.006
Tilg H, Kaser A (2011) Gut microbiome, obesity, and metabolic dysfunction. J Clin Investig 121(6):2126–2132. doi:10.1172/JCI58109
Tremaroli V, Backhed F (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489(7415):242–249. doi:10.1038/nature11552
Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, Nowotny P, Waldhausl W, Marette A, Roden M (2005) Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes 54(9):2674–2684
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI (2009) A core gut microbiome in obese and lean twins. Nature 457(7228):480–484. doi:10.1038/nature07540
Uronis JM, Muhlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C (2009) Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One 4(6):e6026. doi:10.1371/journal.pone.0006026
Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT (2010) Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328(5975):228–231. doi:10.1126/science.1179721
Vital M, Howe AC, Tiedje JM (2014) Revealing the bacterial butyrate synthesis pathways by analyzing (meta) genomic data. MBio. doi:10.1128/mBio.00889-14
Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M (2012) Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143(4):913–916 e917. doi:10.1053/j.gastro.2012.06.031
Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, O’Donnell CJ, Carr SA, Mootha VK, Florez JC, Souza A, Melander O, Clish CB, Gerszten RE (2011) Metabolite profiles and the risk of developing diabetes. Nat Med 17(4):448–453. doi:10.1038/nm.2307
Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ (2006) Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 40(3):235–243
Wu G (2013) Functional amino acids in nutrition and health. Amino Acids 45(3):407–411. doi:10.1007/s00726-013-1500-6
Wu G, Bazer FW, Dai Z, Li D, Wang J, Wu Z (2014) Amino acid nutrition in animals: protein synthesis and beyond. Annu Rev Anim Biosci 2:387–417. doi:10.1146/annurev-animal-022513-114113
Zhang LM, Nichols RG, Correll J, Murray IA, Tanaka N, Smith PB, Hubbard TD, Sebastian A, Albert I, Hatzakis E, Gonzalez FJ, Perdew GH, Patterson AD (2015) Persistent organic pollutants modify gut microbiota-host metabolic homeostasis in mice through aryl hydrocarbon receptor activation. Environ Health Perspect 123(7):679–688. doi:10.1289/ehp.1409055
Acknowledgements
This work was supported by National Natural Science Foundation of China (no. 81600509).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that there is no conflict of interest regarding the publication of this article.
Ethical statements
This review does not involve any human participants and animal work.
Additional information
Handling Editors: C.-A. A. Hu, Y. Yin, Y. Hou, G. Wu, Y. Teng.
Rights and permissions
About this article

Cite this article
Lin, R., Liu, W., Piao, M. et al. A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids 49, 2083–2090 (2017). https://doi.org/10.1007/s00726-017-2493-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-017-2493-3