
Abstract
LC-Q-Orbitrap efficiency was evaluated for pesticide multi-residue analysis by using three workflows involving simultaneous MS and MS2 analysis. They were as follows: data-dependent MS2 (dd-MS2), all-ion fragmentation (AIF) and variable data-independent analysis (vDIA). These MS2 workflows were tested for the main method validation parameters such as detection and identification capabilities, repeatability, linear range and quantitation. QuEChER acetonitrile extracts (blanks and spiked with 166 pesticides) of 11 different fruits and vegetables were used for this evaluation. Blank extracts were analysed to evaluate isobaric compounds and potential false identification. Spiked extracts (at 0.01 and 0.1 mg kg−1) were analysed to evaluate the false negatives potentially produced (considering a retention mass window of 0.2 min). At 0.01 mg kg−1, dd-MS2 had the highest identification rate (96–100%, depending on the matrix). In vDIA, it was 86–100% and in AIF 81–100%. But these two last workflows offered more possibilities for applying screening analysis. It was observed that application of the ion ratio criterion established in the SANTE Guidelines for identification can generate some artificial false negative results. It could be overcome by considering a mass error threshold (i.e. 5 ppm) as a selected criterion. Detection and quantitation were carried out in full-scan MS. MS2 data were used for identification. Dd-MS2 provided the highest number of points per chromatographic peak, and by that peak area, repeatability was the best (typically <10%). AIF and vDIA were characterised by longer cycle times; thus, the obtained peak area repeatability was slightly worse, but acceptable (<20%). All workflows showed very good linearity in the range 0.01–0.5 mg kg−1. The three MS2 workflows were applied to real samples with good results.

LC-Q-Orbitrap was used for pesticide residues analysis in fruit and vegetables. Three approaches to MS2 identificaton were evaluated
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Today, triple quadrupole mass spectrometry (QqQ) is considered as a “gold standard” in pesticide residue analysis. This technique has become the first choice for routine analysis of pesticides in fruits and vegetables. It has great potential in quantitative analysis because of its very high sensitivity in SRM (selected reaction monitoring) and broad linear range. During the last years, the sensitivity and dynamic range of these instruments were intensively developed by the producers. However, the selectivity practically has not changed [1].
On the other hand, high-resolution accurate mass spec-trometry (HRAMS) has improved the qualitative (resolution and mass accuracy) as well as the quantitative (sensitivity and linear range) aspects. In the literature, numerous studies comparing QqQ with HRAMS can be found. The typical conclusion is that HRAMS provides the same or better selectivity than QqQ [2–7]. HRAMS instruments are well known for their high selectivity in full-scan MS mode [8]. Nevertheless, even spectrometers which have very high reso-lution can produce false positive results [9, 10]. Isotopic pat-tern and alternative adducts cannot always correctly discard false positives. For example, it is impossible when a matrix ion has the same elemental composition as a pesticide. A solution to reduce the number of false positive results is the application of simultaneous full-scan MS and MS2. Modern mass spectrometers are hybrid instruments, and by that, it is possible to work with MS2 mode. Besides a HRAMS analyser, they are also equipped with a quadrupole mass filter or at least a collision cell. Even if an instrument does not have a collision cell, fragmentation is possible in the ionisation source [11].
The widest range of opportunities is provided by instruments with quadrupole mass filters. Quadrupoles make available workflows that can be divided into two categories: targeted and non-targeted. In the case of targeted workflows, an inclusion list of precursor masses and retention times is necessary. In this type of workflow, narrow quadrupole isolation windows are recommended. When isotope ions are not a matter of interest, the window can be approx. 1 Da wide. To also obtain fragments of isotopic precursors, a wider isolation window is necessary. MS2 scan can be triggered in the case of detection of the targeted precursor or carried out alternatively with full-scan MS independently of the presence or absence of the precursor from the inclusion list. In the case of non-targeted methods, the quadrupole mass filter stays fully or partially open. When the quadrupole filter is fully open, ions from a whole mass range are fragmented and subsequently analysed by the HRAMS analyser. To increase the selectivity of the method, the entire mass range can be divided into a series of narrower mass ranges which are isolated one by one by the quadrupole and fragmented separately. This approach decreases the number of possible interferences in the MS2 spectrum [10–12] proportionally to the number of mass segments.
In HRAMS, one representative ion is enough for detection. However, a positive identification needs at least one additional ion. It can be an isotope or other adducts (sodium, potassium or ammonium); however, fragment ions are preferable. Adducts and isotopes do not always have enough selectivity. In-source-generated fragments usually have low abundance because when parameters of the ionisation source are being optimized, the main objective is to obtain high sensitivity for the (de)protonated molecule. Therefore, the best solution is identification using MS2 fragment ions. According to the current guidelines of DG SANTE [13], in the case of methods without precursor selection (with a widely open quadrupole), a fragment with mass accuracy of 5 ppm is necessary. The peak of this ion has to overlap with the chromatographic peak of the ion used for detection, and the ion ratio of these two ions has to be within ±30% (relative) of the ratio of the standard. In MS2 workflows with precursor ion selection, there is not any specified mass tolerance.
The main objective of this paper was to compare three workflows of simultaneous MS and MS2: all-ion fragmentation (no precursor selection, ions from the entire mass range fragmented at the same time); variable data-independent acquisition (no precursor selection, mass range divided into smaller segments before fragmentation); and data-dependent MS2 (selection of precursor). This evaluation can facilitate the selection of the optimal workflow and its operational parameters in the field of pesticide residue analysis.
Material and methods
Reagents and materials
High-purity pesticide standards were obtained from Dr. Ehrenstorfer (Augsburg, Germany) and Riedel–de Haën (Selze, Germany) and were stored at −30 °C. Individual pesticide stock solutions (1000–2000 mg L−1) were prepared in acetonitrile and ethyl acetate and were stored in amber screw-capped glass vials in the dark at −20 °C. Individual standard solutions, used for the optimization, along with standard–mix solutions, used for the calibration, were prepared from the stock standards.
Water was obtained from Fisher Scientific (Fair Lawn, NJ, USA) and methanol from Fluka Analytical (Steinheim, Germany). Ammonium formate and formic acid were purchased from Sigma-Aldrich (Steinheim, Germany). Pierce LTQ Velos ESI Positive Ion Calibration Solution was provided by Thermo Fisher Scientific (Waltham, MA, USA).
LC-MS analysis
For the LC separation, UHPLC Dionex™ Ultimate 3000 (Thermo Scientific™, San Jose, CA, USA) was used. Mobile phase A was 98% water and 2% methanol, whereas mobile phase B was 98% methanol and 2% water; both mobile phases contained 5 mM ammonium formate and 0.1% formic acid. Separation was carried out on a Thermo Scientific™ Accucore™ aQ C18 column. The length, diameter and particle size were 100 mm, 2.1 mm and 2.6 μm, respectively. The column was thermostated at 30 °C. The mobile phase gradient started from 100% of mobile phase A and was maintained for 1 min; from 1 to 2 min, the amount of mobile phase B increased to 30%, from 2 to 3 min to 50% and from 3 to 11 min to 100%. One hundred per cent of B was maintained until 14 min. Following this, the mobile phase was changed to 100% A and maintained over 3 min for re-equilibration. The injection volume was 10 μL. The autosampler was thermostated at 10 °C.
A QExactive Focus (Thermo Scientific, Bremen, Germany) mass spectrometer was equipped with a heated electrospray ionisation source (HESI II). The HESI parameters in positive polarity were as follows: sheath gas flow rate, 40; auxiliary gas flow rate, 5; sweep gas flow rate, 1; spray voltage, 3.00 kV; capillary temperature, 280 °C; S-lens RF level, 55.0; heater temperature, 350 °C. In all workflows, parameters of the full-scan analysis were the same: resolution, 70,000; AGC target, 1e6; max, IT auto; scan range, 120–1000. The parameters of data-dependent MS2 were as follows: resolution, 17,500; isolation window, 1.5 m/z; collision energy, 30; AGC target, 5e4; max IT, auto; under fill ratio, 1%. Parameters of MS2 in all-ion fragmentation were as follows: resolution, 70,000; collision energy, 30; AGC target, 1e6; max IT, auto; scan range, m/z 66.7–1000. Parameters of MS2 in variable data-independent analysis were as follows: collision energy, 30; AGC target, 1e6; max IT, auto. In variable data-independent analysis (vDIA), two resolution values were tested: 17,500 and 35,000. Three and five mass segments were evaluated. In the case of three mass segments, the investigated mass range was divided as follows: m/z 120–270, 265–420 and 415–1000. When five mass segments were applied, m/z 120–205, 195–305, 295–405, 395–505 and 495–1000.
The external mass calibration and the quadrupole calibration were carried out daily. For the calibration, a mixture containing n-butylamine, caffeine, Ultramark 1621 and MRFA was used. TraceFinder 4.0 (Thermo Scientific, San Jose, USA) was used for qualitative and quantitative analyses. Automatic detection and quantification was followed up by a manual verification.
Sample preparation
All matrices were extracted according to the following protocol. A portion (10 g) of the homogenized sample was weighed in a 50-mL PTFE centrifuge tube. Then, 10 mL of acetonitrile was added and the samples were shaken in an automatic axial extractor (AGYTAX®, Cirta Lab. S.L., Spain) for 4 min. Afterwards, 4 g MgSO4, 1 g NaCl, 1 g trisodium citrate dihydrate and 0.5 g disodium hydrogen citrate sesquihydrate were added, and it was shaken again in the automatic axial extractor for 4 min. The extract was then centrifuged (3700 rpm) for 5 min. A 5-mL volume of the supernatant was transferred into a 15-mL PTFE centrifuge tube containing 750 mg of MgSO4, 125 mg of PSA and 125 mg of C18. The extract was shaken in a vortex for 30 s and centrifuged again (3700 rpm) for 5 min. The extracts were then transferred to an amber vial and were acidified with 10 μL formic acid 5% per millilitre of extract.
Extracts were spiked with 166 pesticides (the list of pesti-cides with their masses and retention times is shown in Table S1 in the Electronic supplementary material, ESM), all of which are included in the European Union Monitoring Program. Of the blank extract, 100 μL was evaporated under a gentle nitrogen stream and reconstituted with 100 μL of acetonitrile containing appropriate concentrations of the target pesticides. Subsequently, 400 μL of ultrapure water was added to dilute the sample and reduce the percentage of acetonitrile. The final samples contained 0.2 g of matrix per 1 mL. All concentration values which appear in the paper refer to the concentration before dilution.
Results and discussion
Qualitative analysis
Qualitative analysis in target MS2
When working with target analysis, a list of compounds which are searched by the instrument is present. Before fragmentation, the precursor ions of the target compounds are filtered with a very narrow quadrupole window (e.g. 1 Da). The most important advantage of this approach is the high selectivity of the MS2 spectra obtained. However, to set up a target method, the m/z values of the precursor ions have to be known. Additionally, their retention times are also needed. All target compounds can be fragmented with a generic collision energy (e.g. 30 eV). But there is also the possibility of using an optimized collision energy for each analyte. In that case, the MS2 spectra allow a more certain identification. Nevertheless, optimization of the collision energies requires more time than the selection of a generic value. Another disadvantage is that there are no MS2 data for non-target compounds.
Target MS2 analysis was carried out by use of the data-dependent MS2 mode. Resolutions in MS and in MS2 were set the same as in our previous article [10]. Eleven commodities (see experimental) were selected for the evaluation of the method. Blank extracts were fortified with 166 pesticides at two concentration levels, 0.01 and 0.1 mg kg−1. At each concentration level, 1826 results were obtained (11 matrices multiplied by 166 pesticides). The purpose was to search possible false detects/false results. The matrices were selected to be representative of a wide range of difficulties.
In data-dependent MS2 (dd-MS2), strict application of the ion ratio criteria was not feasible. In this workflow, MS2 data were represented only by a single scan; thus, calculation of the ion ratio between the full-scan peak and the MS2 peak was unrealizable. However, the criterion of identification was fulfilled considering the presence of at least two ions (one in full-scan MS and one in MS2) with mass error lower than 5 ppm. At level 0.1 mg kg−1, all selected compounds were detected in full-scan mode. But in two cases, the detected compounds were not identified by MS2, both because of lack of the MS2 scan. The MS2 scans were not obtained for aldicarb in garlic and lufenuron in parsley. Garlic and parsley are considered as highly difficult matrices that provide high ion suppression. At a level of 0.01 mg kg−1, the total number of false negative results was 44. Aldicarb and lufenuron represented 21 of these cases (11 and 10 false negative results, respectively). The main reason was the low response of these pesticide/matrix combinations close to their LODs, which makes the MS2 scan not triggered because of the low abundance of the precursor ion. At concentration levels of 0.02–0.03 mg kg−1, identification was obtained for the majority of the cases. Therefore, applying extract dilution five times (see experimental part) should not be recommendable for these two compounds when high ion suppression is present.
To fulfil the criterion of the ion ratio specified in the DG SANTE guideline, the MS2 collision energies were optimized. Optimization was carried out in the range 10–90 V in 10-V intervals. The optimization allowed obtaining at least two fragment ions per pesticide, and in consequence, it was possible to calculate the ion ratio between two MS2 fragment ions. The ion ratios obtained were stable. Problems with identification were found only in the case of very low sensitive compounds or when the MS2 scan was carried out very far from the peak apex. The obtained percentages of the identified pesticides were similar to the case of the non-optimized collision energy. However, in this approach, the ion ratio criterion was included in the results evaluation.
Qualitative analysis in non-target MS2
In the non-target mode, there is no target list. For fragmentation, a wide quadrupole isolation window is used. Because of that, the obtained MS2 spectra contain a higher number of fragment ions than in the ddMS2. Therefore, a higher number of isobaric ions is expected. The applied collision energies are generic. By that, fragmentation is not optimal. In a few cases, some non-fragmented precursor ions can be found in the MS2 spectrum because the applied energy is too low. One of the advantages of this approach is that the compound retention times or the masses of the precursor ions are not required in the setup of the acquisition method. Other important advantages of this approach are its simplicity of setup and the MS2 data for all compounds present in the sample. There are two possibilities of the realization of non-target MS2. The precursors from the whole analytical range can be fragmented together (all-ion fragmentation) or the analytical range can be divided into smaller mass segments and fragmented separately (vDIA). vDIA needs further optimization.
Optimization of vDIA parameters
Extracts of two matrices were used in the first stage of experiments: tomato, as an example of a commodity containing a relatively low number of co-extracted matrix compounds, and orange, which contained much more co-extractives than the tomato [14, 15]. The main purpose of the experiments was to reveal the influence of resolution and number of mass segments in MS2 on identification and, indirectly, on detection in the vDIA workflow. A resolution of 70,000 in full-scan MS was applied in all cases. Then, the best-performing vDIA method evaluated was used for further comparison with two other investigated workflows: all-ion fragmentation (AIF) and dd-MS2. Three and five mass segments and resolutions of 17,500 and 35,000 were evaluated.
A compound was considered as detected if the retention time drift was lower than 0.2 min (with respect to the standard) and the mass error lower than 5 ppm (compared to the theoretical mass). Detected compounds were considered as identified if a MS2 fragment was detected with a mass error lower than 5 ppm (compared to the experimental database), the deviation of the ratio of chromatographic peaks of the fragment and precursor ion was lower than 30% and if the peaks in MS and MS2 overlapped.
In the spiked tomato extract, the evaluated parameters of vDIA (number of mass segments and resolution in MS) had no influence on detection. All pesticides were detected independently of the number of mass segments selected (three or five) and the resolution (17,500 or 35,000) used in MS2. In orange extract, some small differences in the full-scan MS detection rate between the selected methods were observed. The differences were related to the cycle length. There were four undetected compounds with the longest cycle time method (five mass segments with resolution of 35,000) and two undetected compounds with the shortest (three mass segments with 17,500 resolution). The mentioned results refer to the level of 0.01 mg kg−1; at the level of 0.1 mg kg−1, all compounds were detected. These undetected results were a consequence of the low sensitivity of aldicarb and lufenuron that decreased as a consequence of the ion suppression and also as a consequence of the limitation of the intrascan dynamic range caused by the high number of ions [16] to values <LOQ.
From the point of view of identification, the most efficient method was by selecting five mass segments with a resolution of 35,000. In tomato extract, 99 and 100% of pesticides were identified at levels of 0.01 and 0.1 mg kg−1, respectively. In orange extract, the corresponding results were 90 and 99%. Again, the reason for the loss of identification was low sensitivity.
The results provided by the method with three mass segments and resolution of 17,500 were the worst. In tomato, the percentages of the identified compounds were equal to 93 and 99% (at 0.01 and 0.1 mg kg−1, respectively), whereas in orange these are 79 and 93%. The advantage of the higher resolution and higher number of mass segments was especially significant at the lower concentration level and complex matrices. The reason for that is the high amount of matrix ions very similar to that produced by the target compound. Making the resolution not high enough for their separation and merging in the mass spectrum as a single ion with high mass error (>5 ppm) therefore report a false negative (Fig. 1).

Influence of resolution in MS2. Identification of 0.01 mg/kg of demeton-S-metylsulfoxide in orange extract. Expected mass of fragment ion, 105.0372. a vDIA three MS2 mass segments with a resolution of 17,500. b vDIA three MS2 mass segments with a resolution of 35,000
Considering the results from both matrices, it can be stated that the identification capabilities of two other vDIA settings (three segments at 35,000 and five segments at 17,500) were very similar. Independently, if they were compared at a low or at a high concentration level and in complex or in a simple matrix, the percentages of the identified pesticides were the same or almost the same and worse than the five mass windows at 35,000.
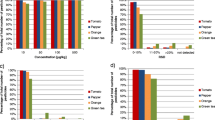
Considering the best identification rate, the settings with five mass segments and resolution of 35,000 were selected for comparison with AIF and dd-MS2. Figure 2 presents the percentages of the identified pesticides with all tested vDIA settings.

Percentages of the identified compounds with various vDIA settings
vDIA results
This evaluation was carried out by using the same matrices and concentration levels as in the case of dd-MS2 (“Qualitative analysis in target MS2 ”).
Compared to dd-MS2, vDIA reported a higher number of false negative results (especially in the case of difficult matrices). Twelve MS2 false negatives were obtained at the level of 0.1 mg kg−1, which was 0.7% of the total number of results. All compounds were detected in full-scan MS, but the problem was lack of identification in MS2. A high number of false negative results was obtained at the level of 0.01 mg kg−1 (6% of the total number of results); amongst them, in 16 cases, the pesticide was not detected in full-scan MS (0.1% of the total number of results). It is important to note that the results described above were obtained by applying the criteria of mass error (<5 ppm), co-elution (0.01 min) and ion ratio variation (±30%). However, evaluating the same results using the criteria of co-elution (0.01 min) and mass error (<5 ppm) for both modes, full-scan MS and MS2, the number of false negative results diminished significantly, from 0.7 to 0.1% at 0.1 mg kg−1 and from 6 to 2% at 0.01 mg kg−1. Thus, the results were very similar to those obtained with dd-MS2. This fact points out the interest to consider mass error in MS and MS2 as an identification criterion.
Ion ratio deviated more than 30% because of the presence of isobaric ions in the matrix. As an example of this problem, the case of methiocarb can be shown. In Fig. 3, the extracted ion chromatograms of methiocarb in full-scan MS and its fragment ion in MS2 (vDIA) are shown. In the solvent and in tomato extract (non-complex matrix), the fragment ion is easy to integrate, although the baseline is high. However, in orange extract, the fragment ion of methiocarb is not separated from a nearby eluting isobaric ion. In this case, it is impossible to integrate correctly the peak of methiocarb fragment ion. The matrix in which the ratio deviation was observed most frequently is parsley. In total, it was observed 21 times, 15 at 0.01 mg kg−1 and six at 0.1 mg kg−1. Parsley does not contain as many co-extracted matrix compounds as leek and garlic; nevertheless, the number is considerably higher than in non-complex matrices such as tomato and lettuce. Other matrices in which the ion ratio problem was encountered frequently were the following: onion, 13; garlic, 11; and orange, 10.

Extracted ion chromatograms of methiocarb and its fragment ion. Data obtained with vDIA
The deviation of the ion ratio was higher than 30% (close to 100%), mostly in the case of low concentration of pesticides and complex matrices. Low concentration levels are more susceptible to ratio deviations because a relatively small amount of isobaric ion can considerably affect the ratio. Some general coincidence between the number of co-extracted matrix compounds and the number of compounds with deviated ratios was observed. More complex extracts contain more ions, so the probability that one of them will generate fragment ions isobaric to the fragment ion of the pesticide is higher even at a resolution of 35,000. The summary of identification records is presented in Fig. 4 (for more detailed information, see ESM Table S2).

Percentages of the identified compounds with dd MS2 (one fragment ion with mass error <5 ppm) (a); vDIA (one fragment ion with mass error <5 ppm and variation of ion ration <30%) (b); and AIF (one fragment ion with mass error <5 ppm and variation of ion ration <30%) (c)
As was demonstrated above, the ion ratio can be irreproducible not necessarily because of the irreproducibility of fragmentation in the collision cell but because of the non-target character of the workflow and strong influence of the matrix.
AIF results
In the case of all-ion fragmentation, the mass range analysed in full-scan MS mode was fragmented in a subsequent scan. Because of the huge amount of MS2 fragment ions, a resolution of 70,000 was selected for MS2.
In terms of MS2 identification, the results obtained with AIF were worse than those from vDIA and dd-MS2. Comparing with vDIA, the percentage of the non-identified compounds at a concentration level of 0.01 mg kg−1 rose from 6 to 10%. It was caused by the lower selectivity of AIF compared with vDIA. In AIF, interfering fragment ion could be generated from any ion from the range m/z 120–1000. In vDIA, possibility of the appearance of interfering ion was lower because of the limited range of precursor ions undergoing fragmentation. An example is shown in Fig. 5.

Improvement of selectivity in vDIA compared to AIF. 0.01 mg/kg of propargite in leek
vDIA provided significantly better results than AIF, especially in a considered difficult matrix such as onion. In this matrix, 15 more compounds were identified (almost 10% of the evaluated pesticides) in vDIA with respect to AIF. Improvement was also noted in garlic (11 compounds) and orange (10 compounds).
False positive results
In all analysed pesticide/commodity combinations, only one potential positive result was found. Triazophos was detected in full-scan MS in a blank orange extract. The mass error was equal to 2.6 ppm and the retention time shift was 0.08 min; thus, the detection criteria were fulfilled. The estimated concentration was 0.01 mg kg−1. However, the dd-MS2 data revealed that the ion detected in full scan was not triazophos. Figure 6a, b presents the dd-MS2 spectrum of 0.01 mg kg−1 of triazophos in orange extract and the dd-MS2 of the compound detected in the blank extract, respectively. None of the triazophos MS2 fragments are present in the spectrum of the compound detected in the blank extract. In the case of the results obtained with vDIA, this MS2 fragment was also present in the extract spiked with 0.01 mg kg−1, but not in the blank extract. Data from AIF are not present in Fig. 6 because 0.01 mg kg−1 was below the level of identification of triazophos in orange extract. Except triazophos in orange extract, no other pesticide in a concentration close or above 0.01 mg kg−1 was detected. This negligible amount of false positives detected is a consequence of the excellent resolution and mass accuracy obtained.

False positives of triazophos. a dd-MS2 spectrum of 0.01 mg/kg of analytical standard of triazophos in orange extract. b dd-MS2 spectrum of a peak detected as triazophos in blank orange extract. c vDIA MS2 fragment ion of triazophos (162.0662 ± 5 ppm) extracted from orange extract spiked with 0.01 mg/kg of analytical standard. d vDIA MS2 fragment ion of triazophos (162.0662 ± 5 ppm) extracted from blank orange extract
Quantitation
Peak area repeatability
The quantitation capabilities of the three MS2 workflows commented above (four settings of vDIA, AIF and dd-MS2) were evaluated by a comparison of the peak area repeatability in full-scan mode (ESM Table S3). Differences in peak area repeatability between the workflows came mainly from the cycle time length. A longer cycle time resulted in lower number of points per chromatographic peak. Full-scan MS data were used for quantitation in all of the investigated methods. The number of points per chromatographic peak depended on the length of full-scan MS and the time which the instrument devoted to the acquisition of MS2 data. Full-scan MS data were registered with a resolution of 70,000 at m/z 200. To obtain this resolution, Q Exactive Focus needed around 0.27 s/scan. In the case of data-dependent MS2, the cycle time was practically equal to the cycle time of the full-scan MS. Only one MS2 scan was carried out when the targeted compound was detected in full scan. MS2 scan was carried out with a resolution of 17,500; thus, it takes only around 0.08 s. In all-ion fragmentation, each full MS scan was followed by a MS2 scan. Both were registered with a resolution of 70,000; thus, theoretically, the cycle time was equal to 0.54 s. In the case of vDIA, the theoretical cycle times were from 0.51 to 0.97 s depending on the resolution and number of mass windows selected.
Peak area repeatability was evaluated at two concentration levels (0.01 and 0.1 mg kg−1) in two representative matrices. In tomato, which contains a relatively small amount of co-extracted matrix compounds, and in orange, which is a complex matrix.
In tomato extract at a concentration level of 0.1 mg kg−1, no differences between the workflows were present. The peaks of all the compounds had RSD below 20% and over 98% had RSD below 5%. Some differences were observed at the level of 0.01 mg kg−1. The obtained RSD values were related to the cycle time length. Worse repeatability was especially noticeable for vDIA with five MS2 mass segments registered with a resolution of 35,000 (method with the longest cycle time). This difference is especially clear looking at the percentage of samples with very low RSD (≤5%). In vDIA, 81% of pesticides obtained repeatability better than 5%, whereas in data-dependent MS2 this is 95%. In general, pesticides in the orange extract showed worse repeatability than in tomato as a consequence of the difficulties commented above. This statement refers especially to the concentration level of 0.01 mg kg−1. vDIA with five segments and resolution of 35,000 showed considerably worse peak area RSD than the other workflows. However, even in orange extract spiked at 0.01 mg kg−1, over 90% of pesticides had peak area RSD below 20%. Pesticides with RSD higher than 20% were those which were characterised by a very low response. The difference in RSD was mainly a consequence of the number of points obtained per peak. vDIA with five mass segments with resolution of 35,000 has considerably longer cycle time than the other methods. In vDIA with five mass segments, the criterion of RSD < 20% was not fulfilled by 16 pesticides, whereas in dd-MS2 only nine. In this point, with dd-MS2, we have to take into account that in samples the number of present compounds will be much lower than that in spiked samples, and by that, the RSD will improve.
Additionally, it was also observed that the cycle time of vDIA or AIF was longer than the simple sum length of the component scans. The cycle was longer by about 15–30% than the sum of the lengths of the scans included in the cycle. For example, a scan with a resolution of 70,000 takes 0.27 s. However, a full cycle in AIF containing two scans with a resolution of 70,000 (one in MS and the other in MS2) takes 0.62 s instead of 0.54 s.
During the studies, a negative effect of the use of lock mass on cycle time was observed. It was noticed that if the method included lock mass, the scan rate was lower in comparison to a method without lock mass. Three different approaches were compared to evaluate the influence of lock mass on the scan rate. The comparison was carried out in vDIA with five mass segments with a resolution of 35,000. In the first approach, lock mass was registered during the whole chromatographic run and, in the second, during 30 s (from RT 2.0 to 2.5 min); in the third approach, lock mass was not registered. Subsequently, the total number of scans (full MS and MS2) registered in 12 min was counted and, finally, an average scan rate was calculated. The following results were obtained: lock mass registered during the whole run, 3.70 spectra/s (2661 spectra in 12 min); lock mass registered during 0.5 min, 3.76 spectra/s (2708 spectra in 12 min); without lock mass, 4.66 spectra/s (3354 spectra in 12 min). It is evident that the use of lock mass slows down the system. And it does not make any difference if the lock mass is registered during the whole run or only during a short period. The repeatability studies described above were also carried out with lock mass, and the obtained results were considerably worse than those obtained without lock mass. This was a consequence of the very low number of points per chromatographic peak.
Good repeatability of peak area is one of the factors that are necessary for reliable quantitation. In the case of triple quadrupoles, 10–15 points per chromatographic peak are required to obtain acceptable RSD values of peak area [17, 18]. However, experiments with vDIA revealed that in the Orbitrap system, much less points per peak are able to provide relatively low values of RSD. In the case of Orbitrap, even four or five points per peak are enough to provide acceptable repeatability. That effect can be explained considering that, in the case of triple quadrupoles, one chromatographic point can represent a very low number of ions. However, in Orbitrap, ions are accumulated in the C-trap before the measurement. Thus, one point represents a much bigger population of ions than in the case of triple quadrupoles. By that, the height of each point of the chromatographic peak is more stable and even a low number of points is sufficient to create a chromatographic peak which is useful for quantitation.
Usually, full-scan MS has enough selectivity for quantitation purposes. However, in some cases, especially in very complex matrices, a high number of isobaric interferences appears. Isobaric interferences are particularly problematic for low concentration levels. This problem can be solved in critical cases by quantitation in MS2. Figure 7 presents the extracted ion chromatogram of thiophanate-methyl in onion extract. Application of a 5-ppm mass tolerance window is not narrow enough to remove interferences. Even a smaller mass tolerance does not solve the problem. However, the peak of the MS2 fragment ion is free from interferences. The fragment is selective enough in both vDIA and AIF MS2 data. This fragment ion was characterised by good peak shape, good repeatability and linearity. The same fragment ion was detected in full scan because fragmentation also occurred in the ionisation source, but the abundance of the fragment was one order of magnitude lower than the abundance of the fragment obtained in the high-energy collision dissociation.

Thiophanate-methyl in onion
Linearity and matrix effects
For all three workflows, the linearity was investigated in the range from 0.01 to 0.5 mg kg−1 (after fivefold dilution, the range was from 0.002 to 0.1 mg L−1). In data processing, weighted linear regression (1/x) was applied. The detector response was considered as linear if individual residuals of the five concentrations selected were below 20%.
Each investigated pesticide showed the same linear range in the three investigated methods. Considering the criteria described above, all the pesticides were linear up to 0.5 mg kg−1. Only lufenuron showed some deviations from the linear response. This compound was characterised by low sensitivity. The problem was probably related to a bad electrospray ionisation in the experimental conditions applied.
Matrix effects were evaluated by a comparison of the slopes of the calibration curves in matrix and in solvent. Matrix effects depended on the complexity of the matrix. Typically, signal suppression was observed. Nevertheless, in some cases, the slope of the calibration curve in matrix had a higher value than that in pure solvent. Pesticides in non-complex matrices (tomato and apple) practically could be quantified using standards in solvent because over 95% of the evaluated compounds showed matrix effects lower than 20%. In orange, it was almost 90%, whereas in onion around 50% (ESM Table S4).
Analysis of proficiency test samples
All three workflows were evaluated by analysis of samples from the European Proficiency Test in Fruits and Vegetables (EUPT-FV-15 (potato), EUPT-FV-16 (pepper), and EUPT-FV-17 (broccoli)). Test samples were homogenates of potato, bell pepper and broccoli. The results obtained with dd-MS2, vDIA and AIF were similar. All pesticides were detected and identified. No false positive results were obtained. All concentrations were in agreement with the mean values published in the final reports from the proficiency tests. Differences between concentrations quantified with QExactive Focus and values from the reports in the majority of the cases were lower than 25%.
The concentration values obtained in all workflows were very consistent. Average differences in the case of all results were below 5% (ESM Table S5). The lowest evaluated concentration was around 0.05 mg kg−1 (acephate in potato and pendimethalin in broccoli). At this level, all workflows had very good performance. Some differences in the results could be expected in the case of compounds with concentrations close to 0.01 mg kg−1.
Conclusions
All three evaluated simultaneous full-scan MS and MS2 workflows have important advantages, but also some weak points that have to be considered to select the optimal workflow for the laboratory. From the point of view of quantitation, the most robust is data-dependent MS2. In this workflow, only a small part of the full-scan MS data is lost because of acquisition of MS2 data. In each second, over three scans are carried out, and so presenting a very good identification and quantification performance. AIF is two times and vDIA over three times slower. Longer cycle times can influence the peak area repeatability. On the other hand, workflows such as AIF and vDIA offer the possibility to quantify in MS2, avoiding the negative effects of matrix isobaric compounds. This fact can be significant in very complex matrices such as onion or leek. In some cases, in such commodities, an instrument even as selective as Orbitrap cannot give peaks free from interferences, and MS2 ions represent a good solution.
With Orbitrap technology, chromatographic peaks constructed with four or five points present surprisingly good RSD values. Thus, requirement of 10–15 points per chromatographic peak does not refer to Orbitrap as a consequence of its characteristic acquisition mode (accumulation of ion before measurement).
All tested workflows assured perfect linearity (except for lufenuron). Injecting fivefold diluted extracts of non-complex matrices could be quantified with standard in solvent in many cases. For correct quantitation of complex matrices (especially alliums), matrix-matched calibration is indispensible.
The cycle time of the combined full-scan MS and MS2 is longer than the simple sum of each of the components. System speed is also slowed down in the case of application of lock mass. Therefore, lock mass is recommendable only in dd-MS2. In AIF and vDIA, external calibration is more recommended.
In terms of identification, dd-MS2 works almost excellently. The only problem is when the precursor ion has very low abundance and the number of co-extractives is very high. It is possible that the system will omit this precursor and no MS2 data will be available. It occurred in our study in a few cases. In AIF and vDIA, more false negative results were obtained than in dd-MS2. However, even in the worst cases (low concentrations and complex matrices), close to 90% of compounds were correctly identified. AIF and vDIA are less selective than dd-MS2; thus, more interfering ions are present in the MS2 spectra. However, a great advantage of AIF and vDIA over dd-MS2 is the non-targeted acquisition of the workflow. In dd-MS2, fragments ions can be obtained only for the compounds from the predefined inclusion list. In AIF and vDIA, no inclusion list is present. Theoretically, fragment ions are obtained for all compounds present in the sample. In the case of dd-MS2, retrospective analysis is limited only to full MS. In AIF and vDIA, the MS2 data can also be used for retrospective analysis. Retrospective analysis can be done by use of data from one chromatographic run. No sample reinjection is necessary. Comparing AIF and vDIA, it has to be concluded that AIF assures better quantitation, but not as good identification as vDIA.
For target multi-residue analysis in a routine laboratory, dd-MS2 seems to be advisable for use as a better compromise because MS2 identification is more reliable. However, if the sample may contain pesticides which are not typically analysed by the laboratory, vDIA is a much better solution.
Mass error and co-elution of fragment ions should be used as criteria for identification in workflows such as vDIA and AIF. The additional criterion of ion ratio can lead to false negative results. In the case of workflows such as dd-MS2 (with only one MS2 scan per chromatographic peak), mass errors in MS and MS2 should be considered as the identification criterion. Using only the criteria of co-elution and mass error, the results obtained with vDIA are similar to those obtained with dd-MS2.
References
Kaufmann A. Combining UHPLC and high-resolution MS: a viable approach for the analysis of complex samples? TrAC Trends Anal Chem. 2014;63:113–28. doi:10.1016/j.trac.2014.06.025.
Borràs S, Kaufmann A, Companyó R. Correlation of precursor and product ions in single-stage high resolution mass spectrometry. A tool for detecting diagnostic ions and improving the precursor elemental composition elucidation. Anal Chim Acta. 2013;772:47–58. doi:10.1016/j.aca.2013.02.012.
Kaufmann A. High mass resolution versus MS/MS. Compr Anal Chem. 2012; 58:169–215. doi:10.1016/B978-0-444-53810-9.00001-8.
Kaufmann A, Butcher P, Maden K, Walker S, Widmer M. Reliability of veterinary drug residue confirmation: high resolution mass spectrometry versus tandem mass spectrometry. Anal Chim Acta. 2015;856:54–67. doi:10.1016/j.aca.2014.11.034.
Kaufmann A, Butcher P, Maden K, Walker S, Widmer M. Determination of nitrofuran and chloramphenicol residues by high resolution mass spectrometry versus tandem quadrupole mass spectrometry. Anal Chim Acta. 2015;862:41–52. doi:10.1016/j.aca.2014.12.036.
Kaufmann A, Butcher P, Maden K, Walker S, Widmer M. Comprehensive comparison of liquid chromatography selectivity as provided by two types of liquid chromatography detectors (high resolution mass spectrometry and tandem mass spectrometry): “where is the crossover point?”. Anal Chim Acta. 2010;673(1):60–72. doi:10.1016/j.aca.2010.05.020.
Kaufmann A, Butcher P, Maden K, Walker S, Widmer M. Quantitative and confirmative performance of liquid chromatography coupled to high-resolution mass spectrometry compared to tandem mass spectrometry. Rapid Commun Mass Spectrom. 2011;25(7):979–92. doi:10.1002/rcm.4952.
Kaufmann A. The current role of high-resolution mass spectrometry in food analysis. Anal Bioanal Chem. 2012;403(5):1233–49. doi:10.1007/s00216-011-5629-4.
Rajski Ł, Gomez-Ramos MM, Fernandez-Alba AR. Large pesticide multiresidue screening method by liquid chromatography-Orbitrap mass spectrometry in full scan mode applied to fruit and vegetables. J Chromatogr A. 2014;1360:119–27. doi:10.1016/j.chroma.2014.07.061.
Gomez-Ramos MM, Rajski Ł, Heinzen H, Fernandez-Alba AR. Liquid chromatography Orbitrap mass spectrometry with simultaneous full scan and tandem MS/MS for highly selective pesticide residue analysis. Anal Bioanal Chem. 2015;407(21):6317–26. doi:10.1007/s00216-015-8709-z.
Lehotay SJ, Sapozhnikova Y, Mol HGJ. Current issues involving screening and identification of chemical contaminants in foods by mass spectrometry. TrAC Trends Anal Chem. 2015;69:62–75. doi:10.1016/j.trac.2015.02.012.
Zomer P, Mol HG. Simultaneous quantitative determination, identification and qualitative screening of pesticides in fruits and vegetables using LC-Q-Orbitrap-MS. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2015;32(10):1628–36. doi:10.1080/19440049.2015.1085652.
SANTE/11945/2015.http://ec.europa.eu/food/plant/pesticides/max_residue_levels/guidelines_en
Gomez-Ramos MM, Ferrer C, Malato O, Aguera A, Fernandez-Alba AR. Liquid chromatography-high-resolution mass spectrometry for pesticide residue analysis in fruit and vegetables: screening and quantitative studies. J Chromatogr A. 2013;1287:24–37. doi:10.1016/j.chroma.2013.02.065.
Gómez-Ramos MdM, Rajski Ł, Lozano A, Fernández-Alba AR. The evaluation of matrix effects inpesticide multi-residue methods via matrix fingerprinting using liquid chromatography electrospray high-resolution mass spectrometry. Anal Methods. 2016;8(23):4664–73. doi:10.1039/c6ay00436a.
Kaufmann A, Walker S. Extension of the Q Orbitrap intrascan dynamic range by using a dedicated customized scan. Rapid Commun Mass Spectrom. 2016;30(8):1087–95. doi:10.1002/rcm.7530.
Henderson W, McIndoe JS. Mass Spectrometry of Inorganic, Coordination and Organometallic Compounds. Chichester: Wiley & Sons Ltd; 2005.
Holcapek M, Jirasko R, Lisa M. Recent developments in liquid chromatography-mass spectrometry and related techniques. J Chromatogr A. 2012;1259:3–15. doi:10.1016/j.chroma.2012.08.072.
Acknowledgements
The authors would like to thank Thermo Fisher Scientific for facilitating QExactive Focus and for technical support. The authors acknowledge funding support from the European Commission, DG SANTE (Specific Agreement No. 7 to Framework Partnership Agreement No. SANTE/2005/FOOD SAFETY/0025-Pesticides in Fruit and Vegetables).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that no conflict of interest.
Additional information
Published in the topical collection Advances in LC-MS/MS Analysis with guest editors Damià Barceló and Mira Petrovic.
Rights and permissions
About this article

Cite this article
Rajski, Ł., del Mar Gómez Ramos, M. & Fernández-Alba, A.R. Evaluation of MS2 workflows in LC-Q-Orbitrap for pesticide multi-residue methods in fruits and vegetables. Anal Bioanal Chem 409, 5389–5400 (2017). https://doi.org/10.1007/s00216-017-0220-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0220-2