
Abstract
X-ray chemical element imaging has the potential to enable fundamental breakthroughs in the understanding of biological systems because chemical element interactions with organelles can be studied at the sub-cellular level. What is the distribution of trace metals in cells? Do some elements accumulate within sub-cellular organelles? What are the chemical species of the elements in these organelles? These are some of the fundamental questions that can be addressed by use of X-ray chemical element imaging with synchrotron radiation beams. For precise location of the distribution of the elements, identification of cellular organelles is required; this can be achieved, after appropriate labelling, by use of fluorescence microscopy. As will be discussed, this approach imposes some limitations on sample preparation. For example, standard immunolabelling procedures strongly modify the distribution of the elements in cells as a result of the chemical fixation and permeabilization steps. Organelle location can, however, be performed, by use of a variety of specific fluorescent dyes or fluorescent proteins, on living cells before cryogenic fixation, enabling preservation of element distribution. This article reviews the methods used for fluorescent organelle labelling and X-ray chemical element imaging and speciation of single cells. Selected cases from our work and from other research groups are presented to illustrate the potential of the combination of the two techniques.

Synchrotron X-ray fluorescence distribution maps of Ca, P and S in yeast cells. Elemental distribution maps (green color scale) were combined with the image of vacuoles labeled with Arg-CMAC (red color scale). The yellow signal of superposed images shows that Ca and P are preferentially located within the vacuole.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Knowledge of the distribution of the chemical elements in organelles may reveal their function in a variety of cellular processes. Element imaging can be achieved by use of several micro-analytical techniques, for example histochemical techniques, radioisotope imaging, magnetic resonance imaging, mass spectrometry imaging, and microprobe X-ray fluorescence imaging [1]. Among these, X-ray microspectrometry methods based on synchrotron radiation have proved their usefulness for element imaging and speciation analysis in eukaryote cells [2, 3]. Micro and nano-SXRF (synchrotron X-ray fluorescence) can be used for imaging element distribution at high spatial resolution (<100 nm) [4–6], and micro-XAS (X-ray absorption spectroscopy) has quite unique capability for direct determination of element speciation in cellular compartments, because it can be used to probe the local structure of the absorbing element with high chemical sensitivity [7, 8]. The increased spatial resolution of these methods achieved in recent years now enables imaging of element distribution within the main cellular organelles, nucleus, Golgi apparatus, endoplasmic reticulum, vacuoles, lysosomes, mitochondria, and chloroplasts. However, identification by optical microscopy before X-ray imaging is usually limited to the larger organelles, i.e. the nucleus, the cytoplasm, and the vacuole. Differential interference contrast microscopy (DIC) has, for example, been used to obtain optical images of the morphology of single cells before their analysis by SXRF [9]. In this work on the function of copper in angiogenesis, DIC enabled not only follow-up of cellular differentiation processes but also location of trace elements in relation to cellular shape and nucleus position. Another example is the use of bright-field microscopy for observation of mice oocytes and early embryos; in this work SXRF imaging revealed the critical modification of Zn distribution during meiotic maturation [10]. Combined use of light microscopy and SXRF imaging has been reported for study of element distributions in marine protists [11–13]. It has also been used to reveal nucleolar accumulation of iron in plant cells [14]. The usefulness of light microscopy is relevant for basic identification but is not sufficient when element location must be addressed at the organelle level. To facilitate comparison of element distribution and organelle location, use of correlative fluorescence microscopy with X-ray element imaging can be performed, as proposed by McRae et al. in 2006 [15]. Despite the growing number of applications in cell biology of synchrotron X-ray microspectrometry, the correlation of element distribution with cellular organelles has only been investigated by a few research teams by use of chloroplast auto-fluorescence [11–13], mitochondrial dyes [16, 17], nuclear dyes [17, 18], or, more recently, green fluorescent proteins (GFPs) targeting a specific organelle, for example the Golgi apparatus [19]. As will be discussed, organelle immunolabelling may greatly modify the distribution of elements in cells, because this procedure involves chemical fixation and permeabilization. Therefore, preservation of the distribution of elements in cells requires use of appropriate cryogenic fixation procedures, avoiding chemical fixation and permeabilization. Organelles can be identified before these cryogenic steps, by use of organelle-specific fluorescent dyes or fluorescent proteins on living cells. The purpose of this article is to review the methods for organelle-specific fluorescent imaging combined with chemical element imaging in single cells. The methods will be described and illustrated with some representative examples of applications.
Cryogenic cell preparation and X-ray element imaging
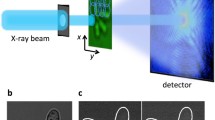
Cryogenic methods of fixation are required to preserve the chemical integrity of the cell and its morphology. In this section we will describe how cells are processed for SXRF and XAS analysis, and how organelle fluorescence can be introduced during the sample-preparation procedure, as summarized in Fig. 1.

Schematic diagram of the procedure for correlative organelle fluorescence microscopy and X-ray chemical element imaging and speciation by use of micro-SXRF and micro-XAS
The first step consists in the cell culture itself, which must be conducted on specific sample holders dedicated to X-ray microanalysis. The sample holder must fulfil at least three important requirements:
-
1.
it must be adapted to the analytical device, which is why, for instance, analysis cannot usually be performed directly on cell culture dishes;
-
2.
the sample holder and the backing must be appropriate for cell culture, meaning it must be non-toxic to cells (Cu grids for electron microscopy should be avoided, for example) and can be prepared under aseptic conditions;
-
3.
the backing must be very pure, containing no detectable trace elements, which is why, for example, glass slides are usually avoided because they may contain traces of elements that can be confused with the cellular elements.
For fluorescent imaging the backing must be transparent, to enable visualization of the cells, and must not interfere with the fluorescence of the dyes. The backing must resist the irradiation and beam damage caused by intense, highly focused X-ray beams. Finally the backing can be in some cases treated with adhesion factors to improve cell attachment. In our experiments, we use a sample holder consisting of a PEEK polymer frame on to which a thin (2 μm) high-purity polycarbonate film is stretched (Fig. 1, top left). Other sample holders, for example ultralene on thin windows, formvar films on carbon coated electron microprobe grids, or thin silicon nitride membranes, can also be used.
Once the cells are in culture on these sample holders they can be processed in the same way as for any cell-imaging method. For instance it is possible to perform immunolabelling during this step but, as will be described in the next section, this procedure will result in extensive redistribution of the elements. The use of live cell imaging with organelle-specific chemical dyes or fluorescent proteins can be performed at this stage, before cryogenic fixation (Fig. 1). Experiments using commercially available organelle fluorescent dyes and the associated procedures will be described in subsequent sections. Before cryofixation, the sample holder is rinsed with buffer and quickly plunged in a cryogenic liquid (for example isopentane, ethane, or propane) cooled by thermal contact with liquid nitrogen at approximately 110 K for isopentane, or ideally at a lower temperature with mixtures of isopentane and propane (80 K), or 77 K with mixtures of ethane and propane. Mixtures of these alkanes have the advantages over pure alkanes of remaining liquid at liquid nitrogen temperature [20]. At this stage samples can be maintained in liquid nitrogen until their analysis, if the sample stage for X-ray spectrometry is equipped with a cryogenic holder. In this case the cells are analysed in a frozen hydrated state, very close to their native state. This is required for speciation analysis of the elements by use of micro-XAS, to avoid any modification of their chemical speciation [16]. This “fully cryogenic” procedure, meaning that cells are cryofixed and then analysed under cryogenic conditions, guarantees preservation of chemical species.
Observing the cells in their frozen-hydrated state is also preferable, to preserve the 3D structure of the cell, and is required for high spatial resolution chemical element imaging. However, SXRF beamlines are not always equipped with a cryogenic sample stage; in this case, therefore, specimens must be freeze-dried after cryofixation, by use of low-temperature and low-vacuum freeze dryers, to preserve both cell morphology and element distribution (Fig. 1). The fluorescence is better preserved at cryogenic temperatures [21], but sometimes the fluorescence is preserved after freeze-drying, enabling observation of the organelles in freeze dried cells at room temperature, which greatly facilitates identification of the cells of interest before SXRF experiments. The persistence of the fluorescence must be checked for each fluorophore and for all experimental conditions. If the fluorescence is not stable sufficiently, the fluorescence microscopy observation is performed on living cells before cryofixation and freeze-drying.
After this sample-preparation procedure, SXRF and XAS can be conducted on single cells. SXRF and XAS methods, and their application to single-cell analysis have been already described in dedicated articles [1–5, 7]. In brief, synchrotron X-ray fluorescence is a multi-element analytical technique that enables determination of the distribution of the elements that compose a specimen. The X-ray beam can be focused at submicron spatial resolution (<100 nm), down to 30 nm for facilities offering the utmost in spatial resolution [5]. A nano-positioner stage moves the sample, and XRF spectra are recorded pixel by pixel which enables imaging of the distribution of the chemical elements, for example in single cells. For micro-XAS the X-ray beam in scanned in energy, around the absorption edge of the element, and the intensity of the beam is measured, before and after passage through the sample, for determination of the absorption coefficient. The spectra used to determine the absorption coefficient are divided into two regions, XANES (X-ray absorption near edge spectroscopy) and EXAFS (extended X-ray absorption fine structure) which provide complementary information about oxidation state and about the chemical nature and bond lengths of the elements. XAS spectrum can be recorded at each pixel position for speciation imaging, but this is a time-consuming modus operandi; usually micro-XAS is performed on selected pixels, for instance within different organelles. If the size of the organelle is larger than the beam size, several point analyses can be performed to analyse the chemical speciation of the entire organelle. This is particularly useful to reveal intra-organelle heterogeneity in chemical speciation, for example within the nucleus [17].
Organelle immunofluorescent labelling: high specificity but important limitations for X-ray element imaging
Immunofluorescence in optical microscopy is important in the life sciences. Its strengths include the specificity of antibody–antigen interactions, the commercial availability of a wide range of primary and secondary antibodies, and the widespread use of microscopes for wide-field and confocal fluorescence imaging. Specific antibodies are available for a large variety of organelles, targeting a specific protein expressed in each organelle. It is, therefore, tempting to consider this technique as a valuable method for correlating the fluorescence microscopy of organelles with SXRF imaging, despite important limitations. Another aspect of the use of antibodies in combination with SXRF is the possibility of detecting them by using the X-ray fluorescence of a metal linked to the antibodies in addition to a fluorescent molecule. Secondary fluorescent antibodies double-labelled with gold particles have been used to detect mitochondria and Golgi apparatus by use of SXRF [6, 15]. Matsuyama et al. [6] reported good correlation between the distribution of Au obtained from SXRF imaging (Au Lα emission) and optical fluorescence microscopy of mitochondria in NIH/3T3 cells after treatment with fluorescein isothiocyanate (FITC), and gold-tagged secondary antibody (Fig. 2a). In this work, element maps (Fig. 2b) were obtained from NIH/3T3 cells fixed with paraformaldehyde (PFA) and treated for immunolabelling. In 2010, the same authors [22] emphasized the need for better control of sample preparation and showed that element distributions are markedly affected by PFA fixation and/or immunolabelling. They compared frozen hydrated cells (Fig. 2c) with cells prepared by use of their previous procedure with PFA fixation and immunolabelling (Fig. 2b). This comparison showed that K is distributed uniformly in frozen hydrated cells whereas it could not be detected in PFA-treated and immunolabelled cells. P and Ca are also present in the whole cell body of frozen hydrated cells and not only in the nucleus, as shown after immunolabelling treatment. The distribution of Fe also depends on the preparation procedure; although predominantly located in the cytoplasm of frozen hydrated cells they are, in contrast, concentrated in the nucleus after use of the PFA–immunolabelling procedure. Cu and Zn are not significantly affected and are found essentially in the nucleus, but also in discrete perinuclear structures, after both procedures. McRae et al. [15] used gold-tagged antibodies to visualize the Golgi apparatus and mitochondrial network by Au SXRF imaging of mice fibroblasts and also noticed some limitations: lower resolution with SXRF than for immunofluorescence microscopy, aspecific binding of Au clusters, and concerns about modification of the concentrations and location of elements depending on the immunolabelling procedure. Indeed, they observed a clear decrease of Ca concentration in cells prepared with permeabilization and washing steps compared with cells prepared with PFA fixation only.

(a) Left: Mitochondrial immunofluorescence of NIH/3T3 cells after treatment with FITC and gold-tagged secondary antibody. The cells were fixed with PFA before being processed by means of a typical immunolabelling procedure. Right: Gold distribution of the same cell imaged by SXRF. (b) Elemental distributions found in PFA/immunolabelled cells: K is not detectable and most of the elements are concentrated in the nucleus; color bars indicate the elemental content (fg). (c) Elemental distributions found in frozen-hydrated NIH/3T3 cells. Ubiquitous distribution of K, P, S, and Ca in the cell body whereas Fe is mainly perinuclear, in contrast with Cu and Zn found predominantly in the nucleus. Scale bars: 10 μm. Figure from original work by Matsuyama et al. [6, 22], reproduced, with permission, from John Wiley and Sons
As illustrated by these studies, the combination of immunolabelling techniques for correlative microscopy with X-ray chemical element imaging has important limitations because of sample preparation. This incompatibility is a consequence of the preparation procedure for immunofluorescence microscopy, which drastically modifies the chemical composition of the sample. The procedure for immunofluorescent labelling of cells grown as monolayers usually involves the following steps:
-
1.
chemical fixation;
-
2.
permeabilization;
-
3.
primary antibody labelling; and
-
4.
secondary antibody labelling [23].
Chemical fixation consists in immobilization of antigens by use of a solution of 2–4 % PFA in phosphate-buffered saline (PBS). When antigens are intracellular, as for organelles, detergents such as triton X-100 or saponin are used to make cell membrane permeable and enable passage of antibodies. After each step cells are rinsed with PBS. This procedure for immunofluorescent labelling of proteins modifies the element distribution within cells compared with cryogenic fixation, as illustrated in Fig. 3.

(a) Live imaging of PC12 cells over-expressing the α-synuclein protein. (b) Same cell after cryofixation and freeze-drying. (c) SXRF imaging, in false-colour (see colour bar), showing the ubiquitous distribution of P, S, and K in the cell body and the organelle-specific distributions of Mn (Golgi apparatus), Fe (cytoplasm), and Zn (nuclei). (d) Optical imaging of PC12 cells over-expressing the α-synuclein protein after processing for immunofluorescence microscopy. (e) Same cell, showing the distribution of α-synuclein immunofluorescence in the cytoplasm (red) and the fluorescent nuclear dye Hoechst 33342 (blue). (f) SXRF imaging of the same cell showing that P, S, K, Mn, Fe, and Zn distributions are altered after immunolabelling. Scale bars: 5 μm
The SXRF experiments presented in Fig. 3 were conducted at the European Synchrotron Radiation Facility (ESRF) on beamline ID22. In this example, in which plunge freezing then freeze-drying were used, the native distribution of the elements was preserved, with P, S, and K being quite homogeneously distributed in the cell, as expected for these ubiquitous biological elements, whereas Mn, Fe, and Zn were located in the Golgi apparatus, the cytoplasm, and the nuclei, respectively (Fig. 3c) [19]. In cells processed for immunofluorescence imaging (Fig. 3d–f) P, S, and K no longer appear homogeneously distributed; the Mn image is essentially background, this element has been fully removed during the sample processing, Fe distribution seems more like a contamination, and Zn is only faintly apparent in the cell. Element loss, i.e. P, S, K, Mn, Fe, and Zn, can be attributed to the permeabilization and rinsing steps, similarly to the observations of Matsuyama et al. for NIH/3T3 cells chemically fixed with PFA and permeabilized with Triton X-100 [6, 22]. Similarly P, S, Cl, and K, losses have been reported after formalin fixation of cerebellum brain tissues compared with cryofixed tissues [24]. We have also observed that PFA and/or solutions of antibodies can contain traces of metals, for example Fe, that could contaminate the preparation. Fe contamination has also been reported in a study of formalin fixation of cerebellum brain tissues [24]. Our qualitative observations are corroborated by the quantitative element analysis presented in Fig. 4.

(a) SXRF spectra of low-Z elements obtained from a single cell after cryofixation and freeze drying (blue) and a cell processed for immunofluorescence microscopy (red). (b) SXRF spectra of high-Z elements obtained from the same single cell. (c) RBS spectra of a single cell after cryofixation and freeze drying (blue), a cell processed for immunofluorescence microscopy (red), and comparison with the signal obtained from the polycarbonate film alone used as backing for the cell culture. The number of counts has been normalized to the number of incoming particles (carbon peak) to enable comparison of the analyses. The procedure for immunolabelling results in large losses of inorganic (S, Cl, K, Mn, Cu, Zn) and organic (C, N, O) elements
In Fig. 4a, b, the number of counts of SXRF spectra has been normalized to the argon (from air) X-ray fluorescence signal to enable comparison of analyses of cells prepared in accordance with the cryogenic or immunolabelling procedures. A drastic loss of elements (P, S, Cl, K, Cu, and Zn) is observed for cells processed for immunolabelling compared with cells processed by cryogenic methods, although Fe content is not modified, probably because loss is counterbalanced by input as a result of exogenous contamination, as suggested by the distribution in Fig. 3f. SXRF measurements indicate that chemical element loss as a result of the immunolabelling procedure is quantitatively very important, especially for such diffusible intracellular ions as K+, large amounts of which are lost because of the permeabilization of the membranes (Fig. 4a). Other elements, for example S and Zn, which are more tightly bound to the proteins are lost, but in smaller proportions (Fig. 4a, b).
Not only are inorganic elements lost; such organic elements as carbon, nitrogen and oxygen are also lost, as is apparent from Rutherford backscattering spectrometric (RBS) analysis (Fig. 4c). RBS is an analytical technique that enables measurement of the organic composition and thickness of such samples as single cells [25]. Incoming particles, protons or alpha particles of MeV energy, are directed on to the sample and the energy loss is measured at a backscattered angle. The energy loss depends on the element with which the interaction occurs and on the depth of this interaction [25, 26]. Knowing the tabulated cross sections for the elements, sample thickness and composition can be determined. The RBS spectrum from the immunostained cells is indicative of large organic element loss compared with cells prepared by the cryofixation procedure (Fig. 4c); the main contribution to the spectrum is from the polycarbonate film. Quantitative analysis of RBS spectra results in areal mass of 70 μg cm−2 for the cryofixed cells and 25 μg cm−2 for the immunostained cells, indicative of 65 % organic mass loss. This is a surprising result, because one could expect that the remaining fixed proteins to contribute a higher proportion to the total areal mass, which is not the case. Soluble cytoplasmic proteins can be lost during the permeabilization process. Depending on the permeabilization conditions, up to 40 % of the total amount of protein can be released [27].
In conclusion, for this section, the procedure for immunofluorescent labelling impedes direct imaging or speciation of chemical elements in immunostained cells. However, if required, immunofluorescence microscopy can be performed in parallel, on a different set of samples, and chemical element mapping can be conducted on untreated samples [28, 29]. For direct correlative microscopy, the procedure for organelle location must be adapted to the cryogenic procedure for element imaging. Organelle fluorescent labelling can be performed on living cells, before the cryofixation step, as described in subsequent sections.
Organelle-specific organic fluorescent dyes
Subcellular fluorescence imaging can be performed easily by use of organic fluorescent dyes that selectively react and associate with organelles. Unlike immunolabelling techniques, these probes have the advantage of suitability for use on living cells. It is thus possible to detect the fluorescent probe without any further chemical fixation and washing steps (i.e. detergent permeabilization) that may substantially alter the trace element content of the cell, as discussed above. A wide variety of fluorescent stains are available commercially (more than 50 are listed in the Molecular Probes Handbook [30]) for all organelles: nucleus, mitochondria, endoplasmic reticulum, lysosomes, Golgi apparatus, etc. Moreover, they cover a wide range of fluorescence and thus facilitate simultaneous multicolour labelling of several organelles. The general mechanism of labelling is based on diffusion across the plasmic membrane (cell-permeable agents) and then a selective localization in organelles according to their reactivity in a specific physicochemical environment. Fluorescent dyes for mitochondria, for example rhodamine, are usually lipophilic cations that accumulate predominantly in mitochondria because of the high membrane potential of this organelle [31]. Retention and accumulation of Lysotracker (Life Technologies) by lysosomes and acidic organelles seems to be because of protonation of the weakly basic dye in low-pH organelles [30]. In subsequent sections we present examples of applications for several organelles and cell types.
Nucleus
Selective accumulation of a fluorescent dye can result not only from a membrane polarization or modification of an acid–base equilibrium but also from selective binding of the dye to a specific component of the organelle. This is especially true for DNA-binding molecules in the nucleus. DAPI (4′,6-diamidino-2-phenylindole), and Hoechst stains such as Hoechst 33258 or 33342 that bind the minor groove of DNA [32, 33] are extensively used for fluorescence imaging of the nucleus. These bis-benzimide dyes bind the minor groove of DNA with high specificity, resulting in blue fluorescence of the nucleus. This staining is, of course, valuable for recognition of the precise location of the nucleus but it can, at the same time, also give precious information about cell-cycle stages (Fig. 5). McRae et al. [18] used this property to correlate the mitotic stages of NIH/3T3 cells with the distribution of trace elements (Fig. 5a). They observed a good correlation between S, Zn, and Cu distribution whereas Fe had a distinct pattern. Another finding was marked variation of Zn concentration during the cell cycle. Direct effects of nuclear fluorescent dyes on trace element distribution have not been observed [18, 19].

(a) Intracellular elemental redistribution in non-synchronized NIH/3T3 cells during mitosis: prophase (Pro.), metaphase (Met.), early anaphase (Early Ana.), late anaphase (Late Ana.), telophase (Tel.), and cytokinesis (Cytokin.). Top row: Fluorescence micrographs of cells stained with Hoechst 33258, a DNA-selective fluorescent probe that highlights the chromosome morphology for assignment of the individual stages of mitosis. 2nd–6th rows: Subcellular distribution of phosphorus (P), sulfur (S), iron (Fe), copper (Cu), and zinc (Zn) for each cell (top row) visualized by SXRF raster scans with excitation at 10 keV and 0.3 μm step size. Scale bars: 10 μm. (b) Intracellular element distributions in interphase NIH/3T3 cells. Top row: Confocal fluorescence micrographs of cells labelled with the cell cycle indicator FUCCI30 for assigning individual interphase stages (red: G1 phase; mixed red and green: G1/S phase; green: G2). 2nd–6th rows: Subcellular distribution of phosphorus (P), sulfur (S), iron (Fe), copper (Cu), and zinc (Zn) for each top row cell visualized by SXRF raster scans with excitation at 10 keV and 0.3 μm spatial resolution. All false-colour maps were normalized to the maximum element density. Scale bars: 20 μm. Figure reproduced from McRae et al. [18], with permission from the Royal Society of Chemistry
Yeast vacuole
Identifying organelles with fluorescent probes has been used in our laboratory for microchemical element imaging of yeast Saccharomyces cerevisiae by use of SXRF. Yeast vacuoles are relatively large organelles approximately 1 to 3 μm in diameter. Living yeasts were incubated for 15 min with the probe Arg-CMAC (7-amino-4-chloromethylcoumarin, l-argine amide, and yeast vacuole marker sample kit (Y7531; Life Technologies) resulting in blue fluorescent staining (λ ex = 354 nm, λ em = 469 nm) of the vacuolar lumen. This chloromethyl coumarin derivative enters the cell easily and is largely sequestrated in the vacuole. It reacts with free thiols of proteins and peptides and its fluorescence is activated after cleavage by vacuolar proteases [30]. This property prevents the washing step necessary to eliminate the excess of fluorescent probe in the medium. After labelling, yeasts were placed on 2-μm polycarbonate foil and excess medium was removed immediately before freezing for 5 min in liquid nitrogen vapour and drying by sublimation at −35 °C for 48 h. Samples were then stored at room temperature in a dry environment until synchrotron radiation analysis. This simple sample preparation procedure was performed because yeast cells do not adhere to the sample holder and cannot be freeze-plunged into a liquid cryogen or placed under vacuum for freeze drying at low temperature and low pressure. Despite this simple processing for cryofixation and freeze drying, the preservation of Arg-CMAC fluorescence after freeze-drying enables precise identification of the freeze-dried yeasts and labelled vacuoles.
SXRF distribution maps of P, S, Cl, K, Ca, and Fe in single yeast cells are presented in Fig. 6. The SXRF experiments were conducted at the ESRF on beamline ID21. Whereas distribution S, Cl, and K was homogenous, and representative of cellular shape, P and Ca were colocalized in the vacuole, as shown by their superimposing distribution with Arg-CMAC. These observations are in agreement with vacuolar storage of P and Ca, because inorganic polyphosphates are able to trap Ca2+ [34, 35].

Chemical element imaging of the yeast S. cerevisiae by use of SXRF. (a) Freeze-dried yeast with Arg-CMAC labelled vacuoles, showing that Arg-CMAC blue fluorescence is preserved after cryofixation and freeze-drying, The Arg-CMAC signal is shown in false colour (red) for the purpose of correlative imaging. (b) SXRF distribution maps of calcium, phosphorus, sulphur, chlorine, potassium, and iron in the same single yeast cell; SXRF imaging was performed by use of the ESRF ID21 beamline with a beam size of 0.4 × 0.7 μm2. (c) Correlative imaging of SXRF chemical element distribution (green—colour scale modified for imaging purposes) and Arg-CMAC signal (red) reveals accumulation of P, Cl, and Ca within the vacuole, as revealed by the yellow colour of the superposed images, not observed for such other elements as S and K
Mitochondria and lysosomes in mammalian cells
Mitochondria are among the most studied subcellular components. These energy-producing organelles are responsible for ATP synthesis by oxidative phosphorylation and lipid oxidation. They are also known to be an important source of radical oxygen species (ROS) and are of crucial importance in apoptosis. Rhodamine 123 is a lipophilic, fluorescent permanent cation that appears yellow–green or red, depending on the properties of the optical filter. This probe is sequestered by active mitochondria in a few minutes. Accumulation of rhodamine by mitochondria is driven by the strong membrane potential (−180 mV) of the inner mitochondrial membrane, which attracts cations to the mitochondrial matrix, and by the lipophilicity of rhodamine, which greatly facilitates crossing of the hydrophobic membranes. We used double labelling of mitochondrial network with rhodamine 123 (λ ex = 507 nm, λ em = 529 nm) and lysosome labelling with Lysotracker blue (L7525; Life Technologies; λ ex = 373 nm, λ em = 422 nm), to investigate arsenic speciation in cells [16, 17]. The micro-SXRF and micro-XANES experiments presented in Fig. 7 were carried out at the ESRF on beamline ID22; a liquid nitrogen cryostream was used to perform both methods at cryogenic temperatures. In this work, lysosomes were labelled after arsenic exposure, cells were incubated with Lysotracker blue (1.25 μmol L−1) in culture medium at 37 °C, with 5 % CO2, for 30 min. Rhodamine 123 (25 μmol L−1 in PBS) was added for 10 min. The live-cells were then washed twice with PBS, pH 7.4, and imaged in less than 20 min under fluorescence microscopy with a water-immersion objective before cryofixation. A predominance of As(III) species was observed in the cytoplasm, mitochondrial network, and lysosomes of HepG2/A cells exposed to As(OH)3 with, in the nucleus, a mixture of trivalent and pentavalent species [17].

SRXF imaging and μXANES subcellular speciation of As in isolated HepG2/A cell. (a) Live cell imaging of HepG2/A cell observed under a light microscope. (b) Same HepG2/A cell with multicolour labelling of mitochondria (rhodamine 123, green) and lysosomes (Lysotracker blue, blue). (c) and (d) SXRF distribution maps of potassium and arsenic. White bars indicate the location and size of the beam during subcellular μXANES measurements. (e) μXANES spectra of subcellular compartments, cytoplasm, mitochondrial network, nucleus, and lysosomes. The dashed line indicates the absorption edge of As(III), the dotted line that of As(V). Use of a liquid nitrogen cryo-stream enabled analysis of the cell in its frozen hydrated state at 150 K, on beamline ID22 at ESRF. Figure reproduced from Ortega et al. y, with permission from Royal Society Publishing
Organelle specific proteins tagged with fluorescent proteins
Green fluorescent proteins (GFPs) can be targeted to a variety of intracellular locations [36, 37]. A method for organelle labelling on the basis of baculovirus gene transfer into mammalian cells, named BacMam technology, is commercially available [38]. BacMam technology for fluorescence imaging involves expression of autofluorescent proteins targeted to specific subcellular organelles. The reagent contains a baculovirus expressing a fusion protein of an organelle marker and a fluorescent protein. BacMam products are available for labelling of organelles including cytoplasm, endoplasmic reticulum, Golgi apparatus, lysosomes, mitochondria, nuclear envelope, nucleus, peroxisome, plasma membrane, and synaptophysin [37, 38].
By use of the BacMam procedure organelles can be located on living cells before cryofixation (Fig. 1) and samples can be further processed in accordance with procedures adapted to SXRF element imaging and speciation. For example, by use of BacMam technology we have shown that manganese accumulates predominantly within the Golgi apparatus of PC12 cells [19]. This specific organelle content was identified owing to the correlation of fluorescence microscopy of GFP-Golgi proteins with SXRF imaging of manganese distribution. We were also able to probe Mn oxidation state in the Golgi apparatus of PC12 cells by using micro-XANES on frozen hydrated cells [19]. Both SXRF and XANES experiments were performed at ESRF on beamline ID21. For Golgi apparatus location, PC12 cells were transduced with Organelle Light reagent (O36215; Invitrogen) expressing the fusion of a Golgi-targeting sequence (N-acetylgalactosaminyl-transferase-2 enzyme) and a fluorescent protein (green, red, or orange; GFP in our work). The procedure was performed, in accordance with the manufacturer’s instructions, 24 h after cells were plated and before Mn addition. Fluorescence of the Golgi apparatus and nucleus labelled with Hoechst stain solution was observed in living cells by use of an epifluorescence microscope before cryofixation. After live cell observation, cells were cryofixed and maintained in liquid nitrogen vapor throughout the experiment, and micro-XANES was performed at cryogenic temperature. The element distribution obtained by SXRF indicated that Mn was located within the Golgi apparatus, and micro-XANES speciation of the oxidation state of Mn revealed it was present as Mn(II) in the cytoplasm and the Golgi apparatus (Fig. 8) [19]. Recently, in a SXRF study of the dynamic distribution of trace elements during cell division, McRae et al. [18] reported the use of this technology (Premo FUCCI cell cycle sensor; Invitrogen) to follow the cell cycle, owing to co-expression of two cell-cycle regulators fused to GFP and red fluorescent protein (RFP). For this correlative comparison, cells were fixed with 3 % PFA after FUCCI sensor expression, observed by confocal fluorescence microscopy, and then prepared for SXRF imaging at the single-cell level (Fig. 5b).

(a) SXRF distribution maps for P, S, and Mn, obtained from frozen hydrated PC12 cells, and fluorescence images of the location of the Golgi apparatus (green, GFP-BacMam) and nucleus (blue, Hoechst stain solution) in live cultured cells. (b) XANES spectra at the Mn absorption edge of the cell presented in (a), within the Golgi apparatus (blue line), and within the cytoplasm (red line), showing that Mn is present in the Mn(II) oxidation state as compared with the Mn(II) standard sample (black line). SXRF and XANES were conducted on hydrated cells cooled by thermal contact with the cryo-stage at liquid nitrogen temperature (77 K), on beamline ID21 at ESRF
Nanoparticle-based methods
Modified nanoparticles can also be used to target organelles. Paunesku et al. [39] developed TiO2 nanoparticles linked to oligonucleotides with a DNA sequence targeting mitochondrial or nucleolar DNA. Several cell lines were transfected with these structures and SXRF confirmed the presence of Ti in the nucleus and mitochondria, depending on the oligonucleotide sequence. A modified version of nanoconjugates with gadolinium attachment—a useful element tag because it is not normally found in cells—has also been investigated by use of SRXF, which revealed possible nuclear accumulation [40]. Later, Thurn et al. [41] reported a method of rendering TiO2–DNA nanocongugates optically fluorescent, enabling direct comparison of the Ti signal from SXRF with images obtained from fluorescent optical microscopy. This kind of modified nanoparticle, initially developed for therapeutic purposes, may be useful for direct SXRF identification of organelles; they are, however, limited to nucleus and mitochondria, and DNA-matching conditions make them highly dependent on cell line species. To target the nucleus, Yuan et al. [42] used nanoparticles of Fe3O4 core and TiO2 shell conjugated to peptides binding the epidermal growth factor receptor (EGFR). High-resolution SXRF imaging and tomography was then conducted to study the distribution of the nanoparticles in the cytoplasm and nucleus. Quantum dots have been used to selectively label cellular organelles [43]; this might have applications in fluorescence and X-ray correlative microscopy of cellular organelles. This principle has been validated with quantum dots–secondary antibody conjugate to label the cancer marker HER2 (human epidermal growth factor receptor 2) on the surface of SKOV3 cancer cells [44].
Conclusion and perspectives
Combination of organelle fluorescence microscopy with SXRF and XAS gives direct information, ideally without sample modification, about the chemical element composition of organelles, and the speciation of these elements. This information will enable better understanding of physiological and toxicological mechanisms involving trace elements at the cellular level, revealing, for instance, nuclear or mitochondrial interactions or detoxification pathways through specific organelles, for example the Golgi apparatus or lysosomes. Fluorescence immunolabelling procedures do not seem to be fully compatible with the X-ray chemical element imaging, because of the large effect of chemical fixation and permeabilization on element concentration and distribution. However, the high specificity and wide range of antibodies available make them a complementary and comparative tool to be applied in parallel to samples different from those analysed by SXRF. Labelling of living cells with organic fluorescent dyes and/or fluorescent proteins, before cryogenic fixation, has been combined with correlative organelle fluorescence microscopy and X-ray chemical imaging. This approach has the advantage of being compatible with live cell imaging, an indication of its relatively low effect on ion homeostasis. Sometimes the fluorescence is stable over time and can be observed even after freeze-drying. However, the best procedures rely on imaging of frozen hydrated cells, because the fluorescence is more stable at cryogenic temperatures and the cellular and organelle structures are better preserved than for freeze-dried samples. This trend is well illustrated by the development of correlative cryo-fluorescence and cryo-electron microscopy [21, 45, 46], and correlative cryo-fluorescence and soft X-ray cryo-microscopy [47–49].
One of the principal objectives of this article is to advocate the more systematic use of organelle fluorescence labelling. Many recent articles reporting SXRF or XAS imaging at the single-cell level would have benefitted substantially from this simple procedure for identification of the organelles in which elements are located. The procedure is also compatible with other chemical element imaging methods, for example electron X-ray microanalysis or micro-PIXE (particle-induced X-ray emission). Another capability that has not yet been fully investigated is the combination of organic fluorophores with different colours and/or expression systems with GFP, RFP, YFP (red and yellow fused proteins) to identify several organelles simultaneously. From the perspective of instrument development, at the time of writing this article, the authors know no of hard X-ray microscope equipped with an epifluorescence microscope enabling correlative imaging on-line. Such implementation, already available for soft X-ray cryo-microscopes [48], would simplify identification of cells and organelles of interest and would result in time saving during X-ray imaging experiments, because only the most interesting cells would be analysed. A distinct and more direct way of obtaining information about element distribution in organelles would be to combine SXRF and high-resolution X-ray microscopy, as has been implemented for soft X-ray microscopy to visualize detailed cellular ultrastructures in 2D [50] and 3D [51], and recently developed for hard X-ray microscopes using phase contrast imaging techniques [5, 52]. Finally, another appealing development would be correlative fluorescence combined with X-ray element 3D microscopy.
References
McRae R, Bagchi P, Sumalekshmy S, Fahrni CJ (2009) In situ imaging of metals in cells and tissues. Chem Rev 109(10):4780–4827
Ortega R, Devès G, Carmona A (2009) Bio-metals imaging and speciation in cells using proton and synchrotron radiation X-ray micro-spectroscopy. J R Soc Interface 6:S649–S658
Vogt S, Ralle M (2013) Opportunities in multidimensional trace metal imaging: taking copper-associated disease research to the next level. Anal Bioanal Chem 405:1809–1820
Carmona A, Cloetens P, Deves G, Bohic S, Ortega R (2008) Nano-imaging of trace metals by synchrotron X-ray fluorescence into dopaminergic single cells and neurite-like Processes. J Anal At Spectrom 23:1083–1088
Chen S, Deng J, Yuan Y, Flachenecker C, Mak R, Hornberger B, Jin Q, Shu D, Lai B, Maser J, Roehrig C, Paunesku T, Gleber SC, Vine DJ, Finney L, VonOsinski J, Bolbat M, Spink I, Chen Z, Steele J, Trapp D, Irwin J, Feser M, Snyder E, Brister K, Jacobsen C, Woloschak G, Vogt S (2014) The Bionanoprobe: hard X-ray fluorescence nanoprobe with cryogenic capabilities. J Synchrotron Rad 21:66–75
Matsuyama S, Shimura M, Mimura H, Fujii M, Yumoto H, Sano Y, Yabashi M, Nishino Y, Tamasaku K, Ishikawad T, Yamauchie K (2009) Trace element mapping of a single cell using a hard x-ray nanobeam focused by a Kirkpatrick-Baez mirror system. X-Ray Spectrom 38:89–94
Ortega R (2011) Direct speciation analysis of inorganic elements in single cells using X-ray absorption spectroscopy. J Anal At Spectrom 26:23–29
Ortega R, Carmona A, Llorens I, Solari PL (2012) X-ray absorption spectroscopy of biological samples. A tutorial. J Anal At Spectrom 27:2054–2065
Finney L, Mandava S, Ursos L, Zhang W, Rodi D, Vogt S, Legnini D, Maser J, Ikpatt F, Olopade OI, Glesne D (2007) X-ray fluorescence microscopy reveals large-scale relocalization and extracellular translocation of cellular copper during angiogenesis. Proc Natl Acad Sci U S A 104(7):2247–2252
Kim AM, Vogt S, O’Halloran TV, Woodruff TK (2010) Zinc availability regulates exit from meiosis in maturing mammalian oocytes. Nat Chem Biol 6(9):674–681
Twining BS, Baines SB, Fisher NS, Maser J, Vogt S, Jacobsen C, Tovar-Sanchez A, Sañudo-Wilhelmy SA (2003) Quantifying trace elements in individual aquatic protist cells with a synchrotron X-ray fluorescence microprobe. Anal Chem 75(15):3806–3816
Twining BS, Baines SB, Vogt S, de Jonge MD (2008) Exploring ocean biogeochemistry by single-cell microprobe analysis of protist elemental composition. J Eukaryot Microbiol 55(3):151–162
Leonardo T, Farhi E, Boisson AM, Vial J, Cloetens P, Bohic S, Rivasseau C (2014) Determination of elemental distribution in green micro-algae using synchrotron radiation nano X-ray fluorescence (SR-nXRF) and electron microscopy techniques–subcellular localization and quantitative imaging of silver and cobalt uptake by Coccomyxa actinabiotis. Metallomics 6(2):316–329
Roschzttardtz H, Grillet L, Isaure MP, Conéjéro G, Ortega R, Curie C, Mari S (2011) The plant cell nucleolus as a hot spot for iron. J Biol Chem 286(32):27863–27866
McRae R, Lai B, Vogt S, Fahrni C (2006) Correlative microXRF and optical immunofluorescence microscopy of adherent cells labeled with ultrasmall gold particles. J Struct Biol 155:22–29
Bacquart T, Devès G, Carmona A, Tucoulou R, Bohic S, Ortega R (2007) Subcellular speciation analysis of trace element oxidation states using synchrotron radiation micro-X-ray absorption near edge structure. Anal Chem 79(19):7353–7359
Bacquart T, Devès G, Ortega R (2010) Direct speciation analysis of arsenic in sub-cellular compartments using micro-X-ray absorption spectroscopy. Environ Res 110(5):413–416
McRae R, Lai B, Fahrni CJ (2013) Subcellular redistribution and mitotic inheritance of transition metals in proliferating mouse fibroblast cells. Metallomics 5(1):52–61
Carmona A, Roudeau S, Perrin L, Veronesi G, Ortega R (2014) Environmental manganese compounds accumulate as Mn(II) within the Golgi apparatus of dopamine cells: relationship between speciation, subcellular distribution, and cytotoxicity. Metallomics 6(4):822–832
Tivol WF, Briegel A, Jensen GJ (2008) An improved cryogen for plunge freezing. Microsc Microanal 14:375–379
Schwartz CL, Sarbash VI, Ataullakhanov FI, McIntosh JR, Nicastro D (2007) Cryo-fluorescence microscopy facilitates correlations between light and cryo-electron microscopy and reduces the rate of photobleaching. J Microsc 227:98–109
Matsuyama S, Shimura M, Fujii M, Maeshima K, Yumoto H, Mimura H, Sano Y, Yabashi M, Nishino Y, Tamasaku K, Ishizaka Y, Ishikawae T, Yamauchia K (2010) Elemental mapping of frozen-hydrated cells with cryo-scanning X-ray fluorescence microscopy. X-Ray Spectrom 39:260–266
Goldstein M, Watkins S (2008) Immunohistochemistry. Curr Protoc Mol Biol 14:14.6
Hackett MJ, McQuillan JA, El-Assaad F, Aitken JB, Levina A, Cohen DD, Siegele R, Carter EA, Grau GE, Hunt NH, Lay PA (2011) Chemical alterations to murine brain tissue induced by formalin fixation: implications for biospectroscopic imaging and mapping studies of disease pathogenesis. Analyst 136(14):2941–2952
Carmona A, Deves G, Ortega R (2008) Quantitative micro-analysis of metal ions in subcellular compartments of cultured dopaminergic cells by combination of three ion beam techniques. Anal Bioanal Chem 390(6):1585–1594
Chu WK, Mayer JW, Nicolet MA (1978) Backscattering spectrometry. Academic Press, New York
Hudder A, Nathanson L, Deutscher MP (2003) Organization of mammalian cytoplasm. Mol Cell Biol 23:9318–9326
Carmona A, Devès G, Roudeau S, Cloetens P, Bohic S, Ortega R (2010) Manganese accumulates within Golgi apparatus in dopaminergic cells as revealed by synchrotron X-Ray fluorescence nano-imaging. ACS Chem Neurosci 1(3):194–203
Yang L, McRae R, Henary MM, Patel R, Lai B, Vogt S, Fahrni CJ (2005) Imaging of the intracellular topography of copper with a fluorescent sensor and by synchrotron x-ray fluorescence microscopy. Proc Natl Acad Sci U S A 102(32):11179–11184
Life Technologies (2010) The Molecular probes® handbook-a guide to fluorescent probes and labelling technologies. 11th edn
Yousif LF, Stewart KM, Kelley SO (2009) Targeting mitochondria with organelle-specific compounds: strategies and applications. Chembiochem 10(12):1939–1950
Portugal J, Waring MJ (1988) Assignment of DNA binding sites for 4′,6-diamidine-2-phenylindole and bisbenzimide (Hoechst 33258). A comparative footprinting study. Biochim Biophys Acta 949(2):158–168
Chen AY, Yu C, Gatto B, Liu LF (1993) DNA minor groove-binding ligands: a different class of mammalian DNA topoisomerase I inhibitors. Proc Natl Acad Sci U S A 90(17):8131–8135
Dunn T, Gable K, Beeler T (1994) Regulation of cellular Ca2+ by yeast vacuoles. J Biol Chem 269(10):7273–7278
Li SC, Kane PM (2009) The yeast lysosome-like vacuole: endpoint and crossroads. Biochim Biophys Acta 1793(4):650–663
Dolman NJ, Kilgore JA, Davidson MW (2013) A review of reagents for fluorescence microscopy of cellular compartments and structures, part I: BacMam labelling and reagents for vesicular structures. Curr Protoc Cytom Chapter 12:Unit 12.30
De Giorgi F, Ahmed Z, Bastianutto C, Brini M, Jouaville LS, Marsault R, Murgia M, Pinton P, Pozzan T, Rizzuto R (1999) Targeting GFP to organelles. Methods Cell Biol 58:75–85
Life Technologies. http://www.lifetechnologies.com/fr/fr/home/life-science/cell-analysis/cell-structure/celllight-ready-to-use-fluorescent-protein-based-reagents.html#table 1 Accessed 12 Feb 2014
Paunesku T, Vogt S, Lai B, Maser J, Stojićević N, Thurn KT, Osipo C, Liu H, Legnini D, Wang Z, Lee C, Woloschak GE (2007) Intracellular distribution of TiO2–DNA oligonucleotide nanoconjugates directed to nucleolus and mitochondria indicates sequence specificity. Nano Lett 7(3):596–601
Paunesku T, Ke T, Dharmakumar R, Mascheri N, Wu A, Lai B, Vogt S, Maser J, Thurn K, Szolc-Kowalska B, Larson A, Bergan RC, Omary R, Li D, Lu ZR, Woloschak GE (2008) Gadolinium-conjugated TiO2–DNA oligonucleotide nanoconjugates show prolonged intracellular retention period and T1-weighted contrast enhancement in magnetic resonance images. Nanomedicine 4(3):201–207
Thurn KT, Paunesku T, Wu A, Brown EM, Lai B, Vogt S, Maser J, Aslam M, Dravid V, Bergan R, Woloschak GE (2009) Labeling TiO2 nanoparticles with dyes for optical fluorescence microscopy and determination of TiO2-DNA nanoconjugate stability. Small 11:1318–1325
Yuan Y, Chen S, Paunesku T, Gleber SC, Liu WC, Doty CB, Mak R, Deng J, Jin Q, Lai B, Brister K, Flachenecker C, Jacobsen C, Vogt S, Woloschak GE (2010) Epidermal growth factor receptor targeted nuclear delivery and high-resolution whole cell X-ray imaging of Fe3O4@TiO2 nanoparticles in cancer cells. ACS Nano 7(12):10502–10517
Delehanty JB, Blanco-Canosa JB, Bradburne CE, Susumu K, Stewart MH, Prasuhn DE, Dawson PE, Medintz IL (2013) Site-specific cellular delivery of quantum dots with chemoselectively-assembled modular peptides. Chem Commun (Camb) 49:7878–7880
Corezzi S, Urbanelli L, Cloetens P, Emiliani C, Helfen L, Bohic S, Elisei F, Fioretto D (2009) Synchrotron-based X-ray fluorescence imaging of human cells labeled with CdSe quantum dots. Anal Biochem 388:33–39
Faas FG, Bárcena M, Agronskaia AV, Gerritsen HC, Moscicka KB, Diebolder CA, van Driel LF, Limpens RW, Bos E, Ravelli RB, Koning RI, Koster AJ (2013) Localization of fluorescently labeled structures in frozen-hydrated samples using integrated light electron microscopy. J Struct Biol 181:283–290
Schorb M, Briggs JA (2014) Correlated cryo-fluorescence and cryo-electron microscopy with high spatial precision and improved sensitivity. Ultramicroscopy 143:24–32
McDermott G, Le GMA, Knoechel CG, Uchida M, Larabell CA (2009) Soft X-ray tomography and cryogenic light microscopy: the cool combination in cellular imaging. Trends Cell Biol 19:587–595
Hagen C, Guttmann P, Klupp B, Werner S, Rehbein S, Mettenleiter TC, Schneider G, Grünewald K (2012) Correlative VIS-fluorescence and soft X-ray cryo-microscopy/tomography of adherent cells. J Struct Biol 177:193–201
Duke EM, Razi M, Weston A, Guttmann P, Werner S, Henzler K, Schneider G, Tooze SA, Collinson LM (2014) Imaging endosomes and autophagosomes in whole mammalian cells using correlative cryo-fluorescence and cryo-soft X-ray microscopy (cryo-CLXM). Ultramicroscopy 143:77–87
Maser J, Osanna A, Wang Y, Jacobsen C, Kirz J, Spector S, Winn B, Tennant D (2000) Soft X-ray microscopy with a cryo scanning transmission X-ray microscope: I. Instrumentation, imaging and spectroscopy. J Microsc 197:68–79
Schneider G, Guttmann P, Heim S, Rehbein S, Mueller F, Nagashima K, Heymann JB, Müller WG, McNally JG (2010) Three-dimensional cellular ultrastructure resolved by X-ray microscopy. Nat Methods 7:985–987
Kosior E, Bohic S, Suhonen H, Ortega R, Devès G, Carmona A, Marchi F, Guillet JF, Cloetens P (2012) Combined use of hard X-ray phase contrast imaging and X-ray fluorescence microscopy for subcellular metal quantification. J Struct Biol 177(2):239–247
Acknowledgments
The authors thank the guest editors N. Jakubowski and P. Dittrich for their kind invitation to publish in this special issue dedicated to “Single Cell Analysis”. In their research the authors have benefited from access to beamlines ID21 and ID22 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We are sincerely grateful to M. Salomé, J. Susini, G. Veronesi, S. Bohic, R. Tucoulou, and P. Cloetens from ESRF and to G. Devès and T. Bacquart from CENBG for providing assistance during the experiments. Some of the developments reported in this review were financially supported by the ANR (Agence Nationale de la Recherche) program PIRIBIO (ANR-09-PIRI-0029-01).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Single Cell Analysis with guest editors Petra Dittrich and Norbert Jakubowski.
Rights and permissions
About this article

Cite this article
Roudeau, S., Carmona, A., Perrin, L. et al. Correlative organelle fluorescence microscopy and synchrotron X-ray chemical element imaging in single cells. Anal Bioanal Chem 406, 6979–6991 (2014). https://doi.org/10.1007/s00216-014-8004-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-8004-4