
Abstract
The application of nanomaterials is leading to innovative developments in industry, agriculture, consumer products, and food and related sectors. However, due to the special properties of these materials there are concerns about their safety, especially because of our limited knowledge of human health effects and the fact that constantly new nanomaterials and applications thereof are being produced. The development of analytical techniques is a key element to understand the benefits as well as the risks of the application of such materials. In this study, a method is developed and validated for sizing and quantifying nano-silver in chicken meat using single particle inductive coupled plasma mass spectrometry (ICP-MS). Samples are processed using an enzymatic digestion followed by dilution of the digest and instrumental analysis of the diluted digest using single particle ICP-MS. Validation of the method in the concentration of 5–25 mg/kg 60-nm silver nanoparticles showed good performance with respect to trueness (98–99 % for size, 91–101 % for concentration), repeatability (<2 % for size, <11 % for concentration), and reproducibility (<6 % for size, <16 % for concentration). The response of the method is linear, and a detection limit as low as 0.1 mg/kg can be obtained. Additional experiments showed that the method is robust and that digests are stable for 3 weeks at 4 °C. Once diluted for single particle ICP-MS analysis, the stability is limited. Finally, it was shown that nano-silver in chicken meat is not stable. Silver nanoparticles dissolved and were transformed into silver sulfide. While this has implications for the form in which nano-silver will be present in real-life meat samples, the developed method will be able to determine the presence and quantity of nanoparticle silver in such samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The application of nanotechnology offers substantial prospects for the development of innovative products and applications, and it is expected that the number of products and production volumes involving nanotechnology will increase in the future. Because of the unique functional properties of nanoparticles (NPs), these materials are being used by many industries, including the food and agricultural sectors [1–6]. While most of these materials are used in the industry, an increasing number of consumer products that contain NPs can already be found on the market. These include electronics, household and cleaning products, paints and coatings, sport products and textiles, cosmetics and personal care products, food products, and food packaging materials. In order to understand how many products containing NPs are already available to consumers, a comprehensive database on types and uses of such NPs would be useful. Since 2006, there is a voluntary database, the Woodrow Wilson Inventory, that is accessible for consumers [7]. More than half of the products in the Woodrow Wilson Inventory contain nano-silver, and although this is by volume not the most used material, it is the fastest growing application of NPs. Nano-silver is used in food supplements, in food packaging as an antimicrobial, in non-food products as toothpaste, and in textiles as an anti-odorant. The silver NPs are added to food packaging materials in several ways, for instance, as finely dispersed nano-silver particles embedded in shrink films, storage boxes, cutting boards containers, and coatings [8–11] (http://factory.dhgate.com/metallized-film/nano-silver-antimicrobial-cling-film-bag-p47593893.html, http://www.gaswatch.com/products/13%22-x-11%22-Antimicrobial-Round-Cutting-Board.html, http://hznano.en.alibaba.com/product/605559706-213764856/Nano_silver_antimicrobial_Storage_Box_manufacturer.html), as a silver-based zeolite in polylactic acid bio-composites [12, 13], and as gelatin-silver nano-composites [14]. Patents also mention the use of silver to prepare antibacterial wheat flour and as an animal feed product [15]. More recently, silver has been studied as an alternative for the antibiotics used in the poultry production [16, 17] and as a nano-agent to control the microbial load in cow milk [18].
Due to the special properties of NPs, there are concerns about their safety, especially because of our limited knowledge of human health effects of these materials and the fact that constantly new NPs and applications thereof are being produced. The development of analytical techniques is a key element to understand the benefits as well as the risks of the application of NPs [19–21]. Currently, separation techniques as hydrodynamic chromatography (HDC) and field flow fractionation (FFF) are used to determine NPs in combination with detectors such as multiple angle light scattering (MALS) and inductive coupled plasma mass spectrometry (ICP-MS) [22–25]. Since detection limits of these combined techniques are in the low milligram per liter range, sample preparation and analyte concentration steps are often required prior to instrumental analysis. However, even fewer methods are available for sample preparation, and those which are available are often difficult and time consuming. We studied a relatively new approach, single particle ICP-MS (sp-ICP-MS), as an innovative method for detection and characterization of silver NPs in chicken meat with limited sample preparation [26, 27]. In addition, sp-ICP-MS is compatible with the recently adopted European Commission recommendation for the definition of nanomaterials, Commission Recommendation 2011/696/EU [28], which states that a particle number-based instead of a mass-based analytical technique should be used. The potential application of silver NPs in the agricultural sector, especially as an antibiotic in poultry production, and in many food-related materials might result in the presence of silver NPs in food. Therefore, silver NPs in chicken meat was selected as target analyte and matrix for the development and validation of an analytical method using sp-ICP-MS as the instrumental technique. Although very recently the extraction and analysis in biological tissues has been described [29], we believe this study to be the first full method validation study using sp-ICP-MS for the sizing and quantification of silver NPs in a food matrix.
Materials and methods
Standards, chemicals, and reagents
Gold NPs, RM8013, is a reference material obtained from NIST (Boulder, CO, USA) and consists of a suspension of gold NPs with a mass concentration of 50 mg/L stabilized in a citrate buffer. These particles have a spherical shape and a diameter of 60 nm. A 50-μg/L stock standard of the 60-nm gold NPs is prepared by diluting 50 μL of RM8013 to 50 mL with Milli-Q water in a glass measuring flask. Stored at room temperature in amber glass screw necked vials, this standard is stable for at least 1 month. Prior to use, the standard is sonicated for 10 min. For the determination of the nebulization efficiency, a 50-ng/L working standard is prepared by diluting 50 μL of the stock standard to 50 mL with Milli-Q water in a 50-mL glass measuring flask. Although this standard is stable at room temperature for several days, it is prepared daily. An ionic silver standard, Ag 100 mg/L, was obtained from Merck (Darmstadt, Germany). Stock standards of 100 μg/L were prepared by diluting 50 μL of the standard to 50 mL with Milli-Q water. Protected from light, this standard is stable at room temperature for at least 2 weeks. Calibration standards in the concentration range of 0.2–5 μg/L were prepared by diluting the ionic stock standard further in Milli-Q water. Protected from light, these standards are stable at room temperature for at least 1 week. Silver NPs, BioPure EAW 1093, were obtained from nanoComposix (San Diego, CA, USA) and consisted of a suspension of silver NPs with a mass concentration of 1,000 mg/L stabilized in a citrate buffer. These particles have a spherical shape and a diameter of 60 nm. A fortification standard of 50 mg/L was prepared by diluting 250 μL of the EAW 1093 to 5 mL with Milli-Q water. Stored at room temperature in amber glass screw necked vials and in the dark, this standard is stable for at least 2 weeks. Prior to use, the standard was placed in an ultrasonic bath for 10 min. Proteinase K, 822 μ/mL, was obtained from Fermentas (Fisher Scientific, Landsmeer, The Netherlands). The digestion buffer consists of 10 mM Tris buffer, 1 % Triton X-100, and 1 mM calcium acetate at pH 9.5. Triton X-100 and calcium acetate monohydrate (Ca(CH3CO2)2 · H2O) were obtained from Sigma-Aldrich (St. Louis, MO, USA), and Tris buffer (hydroxymethyl aminomethane, H2NC(CH2OH)3) was obtained from Merck (Darmstadt, Germany). The digestion buffer is prepared by dissolving 600 mg of Tris buffer and 90 mg of calcium acetate monohydrate in 200 mL of Milli-Q water. Of Triton X-100, 5 mL is added to the solution and mixed with a magnetic stirrer until completely dissolved. This solution is further diluted with Milli-Q water until a final volume of 500 mL. A Milli-Q-Plus ultrapure water system from Millipore (Amsterdam, The Netherlands) was used to obtain high purity water used during sample preparation and dilution of standards and sample suspensions.
Samples and sample processing
The chicken meat for the validation study was purchased from a local supermarket. A 200-mg subsample was collected, cut into small pieces with a surgical knife, and brought into a 10-mL PE tube. The subsample in the tube was fortified with a 50-mg/L aqueous suspension of the 60-nm Ag NPs at 5, 10, and 25 mg/kg. Enzymatic digestion of the sample was carried out in two steps. First, 4 mL of the digestion buffer is added, and the sample is vigorously vortexed for 1 min and sonicated (tip sonication) at 4-W power for 5 min. During sonication, the sample tube is placed in an ice bath to avoid an increase of the sample temperature. Second, 25 μL of proteinase K is added, and the tube is incubated for 3 h at 35 °C. After cooling to room temperature, the digest is diluted 100,000 times and measured using sp-ICP-MS.
Instrumental analysis
The ICP-MS used in this study is a Thermo Scientific X series 2 equipped with a conical glass concentric nebulizer and a quartz impact bead spray chamber. The ICP-MS is operated at a forward power of 1,400 W, and the gas flows were at the following settings: plasma, 13 L/min; nebulizer, 1.1 L/min; and auxiliary, 0.7 L/min. The sample flow rate to the nebulizer was set at 1.0 mL/min using the integrated peristaltic pump. Data acquisition was done using the Thermo PlasmaLab software in the time resolved analysis (TRA) mode. The dwell time was set at 3 ms with total acquisition time of 60 s per measurement. Because of the short dwell time, it is not possible to switch between different m/z values (switching itself takes ~50 ms for the instrument used), and therefore, only one isotope is monitored during the measurement. Isotopes measured for gold (for the determination of the nebulization efficiency) and silver were m/z 197 and 107, respectively. Note that polyatomic interferences are possible, e.g., 91Zr16O for 107Ag; however, considering that the interference is not an NP, it will result in a continuous background signal and can thus be distinguished from the discontinuous NP signals. A typical sample series consists of system blanks (Milli-Q water), a 50-ng/L gold-NP standard to determine the nebulization efficiency, ionic silver standards in the concentration range of 0.2–5 μg/L for calibration, the diluted sample suspensions including method blanks and matrix-matched recovery standards (MMRS, a processed blank sample to which the analyte is added just before the instrumental analyses). The procedure for particle number and particle size calibration is comparable to methods described earlier by Pace et al. [30] and Tuoriniemi et al. [31].
Data processing and calculations
Data are transferred to and processed in Microsoft Excel for the calculation of particle concentrations, particle sizes, and particle size distributions. Acquiring data for 60 s at a dwell time of 3 ms results in 20,000 data points consisting of background signals (ions of the same element, isobaric ions, polyatomic interferences, and instrument noise) and signals with a much higher intensity originating from particles. These particle signals are isolated from the background by plotting a signal distribution, i.e., the frequency with which a signal height occurs as a function of that signal height. This allows a determination of a cutoff point to separate background signals from particle signals (see Fig. 1 for an example). From the number of the particle signals in the time scan and the nebulization efficiency, the particle number concentration in the diluted sample suspension is calculated as follows:

Top left: Time scan of the sp-ICP-MS analyses of a diluted digest of chicken meat containing 60-nm Ag NP at a mass concentration of 10 mg/kg. The Ag NP concentration in the diluted digest itself is ca. 5 ng/L. Left bottom: The signal distribution calculated from the time scan to differentiate between particles and background noise and ions. Right bottom: The particle size distribution that is calculated from the time scan showing a 62-nm Ag NP with a fairly narrow size distribution. Top right: All information about instrument settings, calibration parameters, and measurement results as particle concentrations and size
where C p = particle number concentration (L−1), N p = number of particles detected in the time scan (min−1), η n = nebulization efficiency, and V = sample input flow (mL/min). The same formula is used to calculate the nebulization efficiency after measuring the 50-ng/L 60-nm gold nanoparticle suspension. In that case, the particle number concentration C p is known (50 ng/L of a 60-nm gold particle results in 2 × 107 particles per liter), and the nebulization efficiency is calculated from the observed number of particles N p in the time scan. From the intensity of the particle signals and the response factor calculated from the ionic silver calibration standards, the mass of the individual particles is calculated as follows:
where m p = particle mass (ng), I p = particle signal intensity in the sample (cps), RF ion = ICP-MS response factor from the calibration curve of the ionic silver standards (cps/μg/L), t d = dwell time (s), V = sample flow (mL/min), and η n = nebulization efficiency. To calculate the particle mass concentration in the diluted sample suspension, the masses of all individual particles are summed and corrected for nebulization efficiency and sample flow:
where C m = particle mass concentration (ng/L), m p = particle mass (ng), η n = nebulization efficiency, and V = sample flow (mL/min). Finally, the particle size, expressed as the particle's diameter (and assuming a spherical particle shape), is calculated for each particle as follows:
where d p = particle diameter in the sample (nm), m p = particle mass (ng), and ρ p = particle density (g/mL). The individual particle sizes can be used to produce a size distribution graph. For all these calculations, a calculation template was produced and used.
Electron microscopy
To determine the fate of nano-silver particles after addition to the chicken meat, two digests were studied using scanning electron microscopy (SEM). One digest originated from a sample processed directly after addition of nano-silver, and the other digest was produced 48 h after addition of the 60-nm nano-silver to the sample material. The digests were filtered over nickel-coated polycarbonate filters with 100-nm pores, the filters were rinsed with Milli-Q water to remove salts and soluble organic matter as much as possible, and SEM was used to image the silver particles. The filters were analyzed with high-resolution field emission gun scanning electron microscopy in combination with energy dispersive X-ray analysis (FEG-SEM/EDX). The microscope is a Tescan MIRA-LMH FEG-SEM (Cranberry Twp, PA, USA) operated at an accelerating voltage of 15 kV, working distance 10 mm, spot size 5 nm, and magnification 5.000 to 50.000 times. The EDX spectrometer is a Bruker AXS spectrometer with a Quantax 800 workstation and an XFlash 4010 detector.
Result and discussion
General considerations
Sample pretreatment
Sample preparation techniques are required to isolate NPs from complex media and to prepare a particle suspension suitable for instrumental analysis. Few sample preparation techniques are described in the literature, and what is described is strongly related to the type of NP that is being determined. Helsper et al. describe the isolation of organic NPs in beverages using only aqueous media and physical techniques because organic NPs are essentially micelles that are easily disrupted [32]. On the other hand, Weir et al. used strong acids and elevated temperatures to destroy the sample matrix and isolate relatively inert titanium dioxide particles from food matrices [33]. Ag NPs, however, can undergo many reactions such as dissolution by molecular oxygen and protons [34], reactions with reduced sulfur species and chloride [35, 36], and binding with proteins [37]. To avoid such reactions, care has to be taken during sample preparation, and a relatively soft digestion technique, enzymatic digestion using proteinase K, has been selected for this study. To facilitate the digestion process, the sample is sonicated before the addition of the proteinase K, and the use of chlorine in the enzyme buffer is avoided. Following digestion, the digest is diluted with Milli-Q water for analysis with single particle ICP-MS.
A general problem in method development and validation is the availability of representative materials that actually contain the analyte of interest, i.e., a reference material. Because no reference material or well-defined sample material exists for Ag NPs determination in chicken meat or any other food item, spiked samples are used throughout this validation study. The spikes consist of suspensions of a well-known characterized Ag NPs and are added at the start of the analytical procedure to the sample matrix, generally minced chicken meat. Another point of consideration here is the minimum size of the analytical sample. In a recent article, Linsinger et al. compare the number of NPs per gram of sample to the number of mycotoxin molecules in a sample at the EU maximum residue limit [38]. They concluded that the situation for NPs is comparable with that for molecules and that usual sample sizes are large enough. In addition, they mention that the minimum number of NPs in the sample should be 500 to limit the sampling error of the particle size distribution. Another approach suggested by Peters et al. is to use of Gy's equation to determine the minimum sample size to reach a certain analytical accuracy [39]. Although Gy's sampling theory is hard to digest, theoretical calculations show that a sample size of only 0.01 g of a sample containing 100-nm Ag NPs with a particle mass concentration of 1 mg/kg is sufficient to achieve an analytical accuracy better than 10 %. In addition, a 0.01-g sample of this material contains 10 ng of 100-nm Ag NPs, each having an individual mass of about 5 fg. The number of particles in the sample is thus 2 × 106, far more than the minimum required number of 500 mentioned by Linsinger et al.
Instrumental analyses
The determination of NPs presents analytical challenges different from usual analytical methods, because not only the presence and amount of a substance has to be determined, but also the size of the particles to determine whether it is nano or not. The recent EU definition of nanomaterials complicates things further because it defines a material as a nanomaterial if 50 % or more of the particles in the number size distribution have one or more external dimensions in the size range of 1–100 nm [28]. This means that measuring only size is not enough, but a number-based size distribution has to be determined. Dynamic light scattering (DLS) and electron microscopy (EM) are two methods that can be used to determine number-based size distributions directly. However, DLS produces poor results if other particles or even proteins are present in the sample. Moreover, DLS is not able to determine the identity of the particles and can thus not differentiate between the particles of interest and other particles that may be present in the sample. EM suffers the same problem although identity may be determined if additional techniques as energy dispersive X-ray spectroscopy are used. Nevertheless, a huge number of particles have to be measured, and the preparation of an EM sample may also lead to distortion of the true particle size due to the formation of aggregates. Single particle ICP-MS (sp-ICP-MS) is an alternative that is gaining much interest as a promising new measurement method for the detection and quantification of NPs [40]. In sp-ICP-MS, the sample, an aqueous suspension containing the particles to be analyzed, is introduced continuously into an ICP-MS system that acquires data with a high time resolution. Following nebulization, a fraction of the NPs enter the plasma where they are vaporized and the individual atoms ionized resulting in a cloud of ions. This cloud of ions is sampled by the mass spectrometer and detected as a signal pulse in the detector. A typical run time is 60 s and produces a time scan. The number of pulses detected per second is directly proportional to the particle number concentration in the sample while the intensity of the signal pulse is directly proportional to the mass of the detected nanoparticle. Assuming a certain particle shape (e.g., spheres) and composition (i.e., Ag), one can calculate the diameter of the particle. The method has the advantage of being fast, particle number based and, by virtue of the mass spectrometric detection, very selective. Additionally, it is very sensitive with detection limits in the order of 1 ng/L. Naturally, its applicability is limited to particles of well-defined composition, as otherwise no relation between size and signal exists. The method also shares with basically all methods the feature that it cannot distinguish between single and constituent particles. Nevertheless, since sp-ICP-MS produces a number-based size distribution, it is a useful screening method for the implementation of the EU definition for nanomaterials.
The advantage that most current ICP-MS instruments can be used for sp-ICP-MS is limited by the lack of data processing software. For this purpose, we developed a macro and standard spreadsheet in Microsoft Excel to calculate particle size, particle size distributions, and mass- and number-based particle concentrations. Figure 1 shows a screenshot of the Excel spreadsheet that is used throughout this study.
Method validation
Newly developed methods have to be validated before they can be used in official control studies of foodstuffs. The validation procedure for the determination of residues in foodstuffs from animal origin is described in EU Commission Decision 2002/657/EC. Although the guideline was originally devised for molecular analytes, there is no reason why it should not be used for validation of methods for NP. Following the guideline means that typically over 100 analyses have to be performed for the determination of linearity, CCα, CCβ, trueness, repeatability, and reproducibility. The strategy in this study is to perform seven replicate analyses at three concentration levels on three different days. To determine linearity, a seven-point concentration curve of matrix-matched Ag NP standards is included on each of these days. A scheme of the used validation setup is given in Table 1.
An additional number of samples is analyzed for the determination of selectivity, robustness, and stability. Selectivity is the extent to which other substances interfere with the determination of the analyte, i.e., the Ag NP. To determine selectivity against matrix constituents, a total of 21 blank samples are analyzed by inclusion of seven blank matrices on each validation day. There is no need to determine the selectivity towards other non-silver NPs since the ICP-MS detection itself is highly selective because in sp-ICP-MS the instrument is tuned to only 1 m/z value. Actually, because of the high time resolution, only 1 m/z value can be measured with the currently available ICP-MS instruments. The method is also selective towards the presence of non-nano forms of the same element, e.g., Ag ions, because of the high dilution that has to be applied to observe the single particles. If Ag ions are present in high concentrations, particles can still be observed in the time scan as spikes superimposed on a continuous background signal of ions. The robustness of the method is determined by changing a number of parameters that are believed to affect the measurement results. The variables tested in this study were the sonication time, the digestion time and temperature, and the dilution factor before instrumental analysis. The stability refers to the shelf life of prepared sample materials, digests and diluted digests, and is determined by applying different equilibration times after spiking and re-analyzing materials prepared on the first validation day on the second validation day.
Validation results
Repeatability, reproducibility, and trueness
The repeatability, reproducibility, and trueness are determined by spiking blank samples at three concentration levels, 0.5, 1.0, and 2.5 times the validation level (VL), and analyzing these in sevenfold on three different days that were each more than 1 week apart. VL was set at 10 mg/kg. The measurement data are evaluated for particle diameter and for particle number- and particle mass-based concentration using ANOVA analysis. The results for repeatability, reproducibility, and trueness are given in Table 2.
The results for trueness indicate that the average values that are obtained for the parameters are close to expected values at all three validation levels. The repeatability and reproducibility, both expressed as relative standard deviations, are within 10 % with the exception of the particle number and particle mass concentrations at the 0.5VL level. This is not surprising since in all cases the same dilution factor is used before sp-ICP-MS analyses resulting in lower particle numbers at the 0.5VL level and hence a lower repeatability. Because the NP materials in this study have a relatively narrow size distribution, the average particle diameter does not depend on the number of particles observed; hence, the repeatability and reproducibility are comparable at all validation levels. The relatively large difference between RSDr and RSDRL follows from the fact that the results on different days show larger differences. Apparently, the repeatability of the size measurement is much better than the reproducibility of the preparation of the ionic calibration standards.
Linearity, CCα/CCβ, and LOD/LOQ
The linearity is determined by the analysis of matrix-matched NP standards in the range of 0.05VL–5VL. Although there is some difference in response on the three validation days, all three lines shown in Fig. 2 are linear and show correlation coefficients >0.99. The determination of the repeatability allows for the calculation of the CCα and the CCβ values. CCα, the decision limit, is the concentration above which it can be decided that a maximum residue limit (MRL) has been exceeded with 95 % certainty. CCβ, the detection capability, is the concentration at which MRL concentrations can be detected with 95 % certainty. However, CCα and CCβ are not applicable here since they are calculated based MRL values (MRL = VL) for analytes while for NPs there are no MRLs. Therefore, limits of detection (LOD) and quantification (LOQ) are more appropriate. The LOD and LOQ can be expressed as 3, respectively, ten times the standard deviation of a meaningful blank, i.e., a blank that gives a clearly discernible signal, deviating from a blank showing only noise. The LOD determined in this way corresponds roughly with the “detection capability” CCβ in Decision 2002/657/EC and was estimates from the digests of the 0.05VL matrix-matched NP standard. The average number of peaks in the time scan at this concentration was 52 ± 5 peaks, clearly above the blanks that produced less than ten peaks in any time scan. With a relative standard deviation of 10 %, this means that the LOD is 0.65 mg/kg, and the LOQ is 1.0 mg/kg. With this, it should be kept in mind that a constant dilution factor of 105 was used before sp-ICP-MS analyses of the digest and that the dynamic range of the method can be increased by using dilution factors in the range of 104–106. When a dilution factor of 104 was applied, an LOD and LOQ of 0.07 and 0.1 mg/kg could be reached.

Linearity demonstrated by the analyses of matrix-matched standards of 60-nm Ag NPs in the range of 0.05Vl–5VL on each of the validation days
Robustness, specificity/selectivity
The robustness of the method was tested by varying a number of parameters. These included the sonication time, the time and temperature of the enzymatic digestion, and the dilution factor that was applied before sp-ICP-MS analysis. In all cases, samples spiked at a concentration of 1.0VL were processed in triplicate on one of the validation days. Results were evaluated for particle diameter as well as for particle number and mass concentration. Sonication times of 5, 10, and 15 min were tested, all at a power input of 4 W. Based on the reproducibility in Table 2, the different sonication times did not lead to different results for particle size, number of observed particles in the time scan, and the particle mass concentration. The results are presented in Table 3, and based on these results, a sonication time of 5 min was selected for the final method. The robustness of the method was further tested by changing the digestion time from 1 to 3, 7, and 24 h using a digestion temperature of 37 °C. The results are presented in Table 3 and show that there are no significant effects for each of the parameters. While the results indicate that a digestion time of 1 h is sufficient, a digestion time of 3 h was selected for the final method to include the possibility that differences in meat structures may require longer digestion times. Finally, digestion was carried out at temperatures of 27, 37, and 47 °C, and the results are presented in Table 3. As before, no differences were observed for the particle diameter which ranged from 61 to 62 nm with a standard deviation about 0.5 nm in all cases. However, the particle mass concentration shows a slight decrease from 11.5 mg/kg at 27 °C to 9.0 mg/kg at 47 °C, and a temperature of 37 °C was selected for the final method.
The influence of the dilution factor was tested by diluting a digest of a 1.0VL sample 1,000–100,000 times. The particle size, the number of particles in the time scan, and the corresponding particle mass concentration were compared, and the results in Table 4 show that there is no significant matrix effect between the different dilutions. If the number of particles in the time scan are plotted against the reciprocal dilution a linear relationship is found with a correlation coefficient >0.99.
Selectivity was determined by the analysis of 21 blank samples of chicken meat. In all cases, the number of peaks that were observed in the time scan was <5, comparable for what is found in Milli-Q water. In addition, these peaks show particle diameters that are significantly smaller than the 60-nm particles used in this study. When the developed Excel spreadsheet is used, the particle mass concentrations are below the method LOD for all 21 blank samples. Therefore, it is concluded that the method is specific and selective.
Stability
Stability is a special issue which is often not determined or determined in time and refers to the stability of samples and sample extracts. In this study, the stability at three different stages was determined: the stability of sample digests, the stability of diluted sample digests, i.e., diluted to the level of the sp-ICP-MS analyses, and the stability of spiked sample materials. The stability was tested with digests and diluted digests of chicken meat samples spiked at 0.5VL, 1.0VL, and 2.5VL on the first validation day and stored in the dark for 3 weeks at 4 °C. These stored materials were re-analyzed with the fresh sample digests prepared on the second validation day. Comparison of the results shows clear differences for the fresh and stored digests and diluted digests as presented in Table 5. After 3 weeks, the average particle diameter of the Ag NPs in the sample digests ranges from 60 to 63 nm which is within the reproducibility standard deviation of the method. In the same digests, the recovery of the particle mass concentration ranges from 79 to 85 %, and taking into account the reproducibility standard deviation given in Table 2, this also does not indicate a significant change. Therefore, it is concluded that Ag NPs are stable in the non-diluted digests for at least 3 weeks. In the diluted sample extracts stored for 3 weeks, the particle diameter of the Ag NPs decreased from 60 to 51 nm, a difference well outside the reproducibility standard deviation indicating a significant change. In the same diluted digests, the mass-based particle concentration decreased to 22 % at 0.5VL and to only 8 % at 2.5VL. If the decrease in particle size is a consequence of particle dissolution, then this can explain a decrease of 40 % of the mass-based particle concentration. Additionally, adsorption processes will play a role since NP standards at these low concentrations also show a limited stability. From these results, it is concluded that diluted digests are not stable and should be analyzed as quickly as possible, preferably on the same day they were prepared.
Finally, the stability of the spiked sample material itself was tested. This was undertaken since the preparation of a spiked test material for future use was not successful. Experiments were carried out with sample materials spiked at 10 mg/kg with 60-nm Ag NPs that were either processed directly after addition of the spike or processed after intervals of 2, 24, and 48 h. Spiked materials were stored in the dark at 4 °C until processing. All experiments were carried out in triplicate, and the particle size and particle mass concentration were determined and evaluated. The results as presented in Table 6 show that the particle size decreased from 62 ± 1 nm for particles in samples that were processed directly to 60 ± 2 nm for particles in samples that were processed after 24 h of storage to 56 ± 2 nm for particles in samples processed after 48 h of storage. The results for 24 h of storage are in between, indicating that particles are dissolving after addition to the matrix. This should of course result in an increase of Ag ions in the sample; however, because of the high dilution factor in sp-ICP-MS, this increase in Ag ion concentration will not be observed.
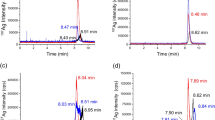
More spectacular is the decrease in particle mass concentration. This drops from 12.1 ± 0.8 mg/kg when directly processed to 4.9 ± 1.1 mg/kg when processed after an equilibrium time of 48 h. This decrease cannot be explained completely by dissolving particles because the change in particle diameter from 62 to 56 nm can explain a maximum mass loss of about 30 %. Another explanation could be the formation of insoluble silver salts like AgCl or Ag2S since chlorine as well as sulfur are expected to be abundantly available in chicken meat. These insoluble materials can possibly agglomerate or deposit and will not be detected in the chemical analysis. To determine what happens to the particles, digests of samples that were processed directly and after 48 h were studied using scanning electron microscopy (SEM) with energy dispersive X-ray spectrometry (EDX) to determine size as well as identity of particles present in the material. Digests were filtered on nickel-coated polycarbonate filters to isolate the particles, and SEM was used to image the silver particles. Figure 3 shows SEM pictures and EDX spectra of particles in both digests. From the SEM pictures in Fig. 3, it is clear that the digest of the sample processed after 48 h contains many less Ag NPs than the digest that was directly processed. Further analysis showed that particles after 48 h were indeed smaller than the spiked Ag NPs, on the average particle size decreased from 60 ± 10 to 50 ± 10 nm. In addition, the EDX spectra of particles in Fig. 3 show that some particles have undergone a chemical transformation from pure silver in the directly processed sample digests to silver sulfide in the digest of samples that were processed after 48 h. It is not clear whether these silver sulfide particles are newly formed via dissolved Ag or these silver particles are in situ transformed into silver sulfide particles. The observation of agglomerates of silver sulfide particles, see Fig. 4, and silver particles that are partly transformed into silver sulfide suggests the latter.

EM pictures and EDX spectra of chicken digests prepared after equilibrium times of 0 and 48 h. The EM pictures show that fewer and smaller particles are present if spiked samples are processed after an equilibrium time of 48 h. The EDX spectra show that pure silver is (partly) transformed into silver sulfide

EM/EDX scans showing an agglomerate in the chicken digest (left), the EDX scan for silver (middle), and the EDX scan for sulfur (right) showing that the particles in the agglomerate actually consist of silver sulfide
It is clear that these observations will have consequences for the form of nano-silver in real-life samples. If silver NPs migrate into meat or if animals are exposed to these particles and uptake takes place, part of this nano-silver may dissolve or be transformed into silver sulfide. The remaining silver NPs, however, will still be detected by the sp-ICP-MS method. Even if silver sulfide is present in the form of NPs, these will also be detected by the sp-ICP-MS method. Because the sp-ICP-MS method quantifies only silver, this may result in an underestimation of the true size of the silver sulfide particle unless it is known that the particles consist of silver sulfide in which case size estimation can be correct.
Conclusions
A method is developed and validated for sizing and quantification of nano-silver in chicken meat using single particle ICP-MS. The choices for sample preparation, instrumental analysis, and validation have been discussed and led to the development of a method using an enzymatic digestion of 200-mg sample material, subsequent dilution of the digest, and instrumental analysis of the diluted digest using single particle ICP-MS. The method was validated following the EU Commission Decision 2002/657/EC for the validation of residues in foodstuffs from animal origin, performing multiple analysis at three concentration levels on three different days. Additional experiments were carried out to determine robustness and stability of the method. The trueness of the method ranged from 98 to 99 % for particle size and from 91 to 101 % for concentration at levels from 5 to 25 mg/kg of 60-nm nano-silver particles in chicken meat. The repeatability ranged from 0.8 to 1.8 % for particle size and 6.7 to 11 % for particle mass concentration. The within-lab reproducibility ranged from 5.0 to 5.6 % for particle size and 8.9 to 16 % for particle mass concentration. The linearity was determined using matrix-matched nanoparticle standards, and the method was found to be linear in the range of 0.5–50 mg/kg of 60-nm nano-silver particles in chicken meat. The LOD of the method can be as low as 0.05 mg/kg. The method is selective, and analysis of blank samples showed that generally <5 particles are identified in blank chicken meat samples. The robustness was tested changing sonication and digestion time, digestion temperature, and dilution factor. The method was robust for these changes with the exception of the digestion temperature for which the highest temperature leads to a lower recovery. Stability experiments showed that sample digests are stable for 3 weeks if stored at 4 °C but that diluted digests are not stable. Finally, it was shown that the 60-nm nano-silver particles spiked to chicken meat are not stable. The particle size as well as the particle mass concentration decreased within 24 h after spiking of the sample material. A study using electron microscopy showed that silver particles dissolved and were chemically transformed into silver sulfide. While this clearly has implications for the form in which nano-silver will be present in real-life meat samples, the developed method will be able to determine the presence and quantity of nanoparticle silver in such samples.
References
Chen H, Weiss J, Shahidi F (2006) Nanotechnology in nutraceuticals and functional foods. Food Technol 3:30–39
Weiss J, Takhistov P, McClements DJ (2006) Functional materials in food nanotechnology. J Food Sci 71:R107–R116
Klaine SJ, Alvarez PJJ, Batley GE, Fernandes TF, Handy RD, Lyon DY, Mahendra S, McLaughlin MJ, Lead JR (2008) Nanomaterials in the environment: behaviour, fate, bioavailability and effects. Environ Toxicol Chem 27:1825–1851
Tiede K, Boxall AB, Tear SP, Lewis J, David H, Hasselhov M (2009) Detection and characterization of engineered nanoparticles in food and the environment–a review. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 7:795–805
Gruére GP (2011) Labeling nano-enabled consumer products. Nano Today 6:117–121
Peters R, ten Dam G, Bouwmeester H, Helsper H, Allmaier G, von der Kammer F, Ramsch R, Solans C, Tomaniova M, Hajslova J, Weigel H (2011) Identification and characterisation of organic nanoparticles in food. Trends Anal Chem 30:100–112
Woodrow Wilson International Center for Scholars (2012) Project on emerging nanotechnologies. An inventory of nanotechnology-based consumer products currently on the market. Washington, DC: Available from: http://www.nanotechproject.org/inventories/consumer/
Benelmekki M, Torrell M, Xuriguera E, Vaz F, Teixeira V (2012) Structure and properties of silver clusters implanted in PET by PVD sputtering for active packaging applications. J Nano Res 18–19:105–116
Martínez-Abad A, Sánchez G, Lagaron JM, Ocio MJ (2012) Development and characterization of silver-based antimicrobial ethylene-vinyl alcohol copolymer (EVOH) films for food-packaging applications. J Agric Food Chem 60:5350–5359
Guo L, Yuan W, Lu Z, Li CM (2013) Polymer/nanosilver composite coatings for antibacterial applications. Colloids Surf A Physicochem Eng Asp 439:69–83
Llorens E, Llorens A, Lloret E, Picouet PA, Trbojevich R, Fernandez A (2012) Metallic-based micro and nanocomposites in food contact materials and active food packaging. Trends Food Sci Technol 24:19–29
Busolo MA, Fernandez P, Ocio MJ, Lagaron JM (2010) Novel silver-based nanoclay as an antimicrobial in polylactic acid food packaging coatings. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 27:1617–1626
Fernandez A, Soriano E, Hernandez-Munoz P, Gavara R (2010) Migration of antimicrobial silver from composites of polylactide with silver zeolites. J Food Sci 75:E186–E193
Halder D, Mitra A, Bag S, Raychaudhuri U, Chakraborty R (2011) Study on gelatin-silver nanoparticle composite towards the development of bio-based antimicrobial film. J Nanosci Nanotechnol 11:10374–10378
Park JY, Li SFY, Kricka LJ (2006) Nanotechnologic nutraceuticals: nurturing or nefarious? Clin Chem 52:331–332
Pineda L, Chwalibog A, Sawosz E, Lauridsen C, Engberg R, Elnif J, Hotowy A, Sawosz F, Gao Y, Ali A, Moghaddam HS (2012) Effect of silver nanoparticles on growth performance, metabolism and microbial profile of broiler chickens. Arch Anim Nutr 66:416–429
Pineda L, Sawosz E, Lauridsen C, Engberg RM, Elnif J, Hotowy A, Sawosz F, Chwalibog A (2012) Influence of in ovo injection and subsequent provision of silver nanoparticles on growth performance, microbial profile, and immune status of broiler chickens. Open Access Anim Physiol 4:1–8
Yildiz N, Pala A (2012) Effects of small-diameter silver nanoparticles on microbial load in cow milk. J Dairy Sci 95:1119–1127
SCENHIR (2007) Opinion on the scientific aspects of the existing and proposed definitions relating to products of nanoscience and nanotechnologies, November 29, 2007 http://ec.europa.eu/health/ph_risk/committees/04_scenihr/docs/scenihr_o_012.pdf
EFSA (2009) Scientific opinion on the potential risks arising from nanoscience and nanotechnologies on food and feed safety, February 10, 2009 http://www.efsa.europa.eu/de/scdocs/doc/958.pdf
Richman EK, Hutchison JE (2009) The nanomaterial characterization bottleneck. ACS Nano 3:2441–2446
Hassellöv M, Readman JW, Ranville J, Tiede K (2008) Nanoparticle analysis and characterization methodologies in environmental risk assessment of engineered nanoparticles. Ecotoxicology 17:344–361
Von der Kammer F, Legros S, Hofmann T, Larsen EH, Loeschner K (2011) Separation and characterization of nanoparticles in complex food and environmental samples by field-flow fractionation. Trends Anal Chem 30:425–450
Von der Kammer F, Ferguson PL, Holden PA, Masion A, Rogers KR, Klaine SJ, Koelmans AA, Horne N, Unrine JM (2012) Analysis of nanomaterials in complex matrices (environmental and biota): general considerations and conceptual case studies. Environ Technol Chem 31:32–49
Dekkers S, Krystek P, Peters RJB, Lankveld DPK, Bokkers BGH, Van Hoeven-Arentzen PH, Bouwmeester H, Oomen AG (2011) Presence and risks of nanosilica in food products. Nanotoxicology 5:393–405
Degueldre C, Favalger PY, Wold S (2006) Gold colloid analysis by inductively coupled plasma-mass spectrometry in a single particle mode. Anal Chim Acta 555:263–268
Laborda F, Lamana JJ, Bolea E, Castillo JR (2011) Selective identification, characterization and determination of dissolved silver(I) and silver nanoparticles based on single particle detection by inductively coupled plasma mass spectrometry. J Anal At Spectrom 26:1362–1371
European Commission (2011) Commission recommendation of 18 October 2011 on the definition of nanomaterial. 2011/696/EU. European Commission, Luxembourg
Gray EP, Coleman JG, Bednar AJ, Kennedy AJ, Ranville JF, Higgins CP (2013) Extraction and analysis of silver and gold nanoparticles from biological tissues using single particle inductively coupled plasma mass spectrometry. Environ Sci Technol. doi:10.1021/es403558c
Pace HE, Rogers NJ, Jarolimek C, Coleman VE, Higgins CP, Ranville JF (2011) Determining transport efficiency for the purpose of counting and sizing nanoparticles via single particle inductively coupled plasma mass spectrometry. Anal Chem 83:9361–9369
Tuoriniemi J, Cornelis G, Hasselhov M (2012) Size discrimination and detection capabilities of single-particle ICPMS for environmental analysis of silver nanoparticles. Anal Chem 84:3965–3972
Helsper JP, Peters RJ, Brouwer L, Weigel S (2013) Characterisation and quantification of liposome-type nanoparticles in a beverage matrix using hydrodynamic chromatography and MALDI-TOF mass spectrometry. Anal Bioanal Chem 405:1181–1189
Weir A, Westerhoff P, Fabricius L, Hristovski K, von Goetz N (2012) Titanium dioxide nanoparticles in food and personal care products. Environ Sci Technol 46:2242–2250
Liu J, Hurt RH (2010) Ion release kinetics and particle persistence in aqueous nano-silver colloids. Environ Sci Technol 44:2169–2175
Liu J, Pennell KG, Hurt RH (2011) Kinetics and mechanisms of nano-silver oxysulfidation. Environ Sci Technol 45:7345–7353
Kaegi R, Voegelin A, Sinnet B, Zuleeg S, Hagendorfer H, Burkhardt M, Siegrist H (2011) Behavior of metallic silver nanoparticles in a pilot wastewater treatment plant. Environ Sci Technol 45:3902–3908
Ravindran A, Singh A, Raichur AM, Chandrasekaran N, Mukherjee A (2010) Studies on interaction of colloidal Ag nanoparticles with bovine serum albumin (BSA). Colloids Surf B 76:32–37
Linsinger TP, Chaudhry Q, Dehalu V, Delahaut P, Dudkiewicz A, Grombe R, von der Kammer F, Larsen EH, Legros S, Loeschner K, Peters R, Ramsch R, Roebben G, Tiede K, Weigel S (2013) Validation of methods for the detection and quantification of engineered nanoparticles in food. Food Chem 138:1959–66
Peters R, Helsper H, Weigel S (2011) NanoLyse EU project. nanoparticles in food: analytical methods for detection and characterisation. Delivarable D4.1a. Sampling of nanoparticles: Relation between sample size and sampling error. http://www.nanolyse.eu/default.aspx
Ranville JF, Reed RB, Mitrano D, Pace H, Higgins CP (2012) Analysis of metal-containing nanoparticles using single particle ICP-MS in environmental matrices, PITTCON 2012, March 12, 2012, Orlando, USA, 11th–15th March 2012
Acknowledgments
This research was commissioned and financed by The Netherlands Food and Consumer Product Safety Authority (NVWA) and The Netherlands Ministry of Economic Affairs (EZ) through its policy support programme (BO).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Characterisation of Nanomaterials in Biological Samples with guest editors Heidi Goenaga-Infante and Erik H. Larsen.
Rights and permissions
About this article
Cite this article
Peters, R.J.B., Rivera, Z.H., van Bemmel, G. et al. Development and validation of single particle ICP-MS for sizing and quantitative determination of nano-silver in chicken meat. Anal Bioanal Chem 406, 3875–3885 (2014). https://doi.org/10.1007/s00216-013-7571-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-7571-0