
Abstract
This publication describes the first international intercomparison of particle-size determination by single-particle inductively coupled plasma mass spectrometry (sp-ICPMS). Concentrated monodisperse silver nanoparticle suspensions with particle diameters of 20, 40 and 100 nm and a blank solution were sent to 23 laboratories in Europe, the USA and Canada. Laboratories prepared eight nanoparticle preparations in two food simulants (distilled water; 10 % ethanol) and reported median particle size, Ag particle mass concentration and Ag particle number concentrations. Average repeatability and reproducibility standard deviation (s r and s R) for the median particle diameter were 1 and 14 nm, respectively. Relative precision was worse for Ag particle number concentrations (RSD r = 11 %; RSD R = 78 %). While further improvements of the method, especially with respect to software tools for evaluation, hardware options for shorter dwell times, calibration standards for determining nebuliser efficiency and further experience by laboratories are certainly desirable, the results of this study demonstrate the suitability of sp-ICPMS for the detection and quantification of certain kinds of nanoparticles.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Recent years have seen an increase in the use of nanoparticles (NPs) in various fields. While the potential benefits of such particles are beyond doubt, concerns have been raised with respect to potential hazardous effects. Legislation requiring the labelling of the presence of materials in their nano-form in cosmetics [1] and food [2] exists on the EU level. To avoid a multitude of conflicting definitions of “nanomaterial”, the European Commission (EC) has published a recommendation for a definition [3] that it intends to use in future laws. This definition defines a nanomaterial as “natural, incidental or manufactured material containing particles, in an unbound state or as an aggregate or as an agglomerate and where, for 50 % or more of the particles in the number size distribution, one or more external dimensions is in the size range 1 nm–100 nm” [3]. Implementation of this definition requires the availability of reliable measurement methods that are able to determine number-based particle-size distributions. A recent report concluded that “none of the currently available methods can determine for all kinds of potential nanomaterials whether they fulfil the definition or not” [4]. Current techniques for the size determination of NPs include fast ensemble methods, such as dynamic light scattering (DLS), which gives intensity-based size distribution, or rather time-consuming counting methods, such as electron microscopy (EM), which does deliver number-based distribution albeit at often prohibitive costs. Consequently, improvement of current methods and development of new methods for the implementation of the definition are therefore paramount.
The concept of utilising ICPMS for single-particle analysis and colloid suspensions was first published by McCarthy and Degueldre [5] and tested for a series of particles in aqueous suspensions including TiO2 and Al2O3 [6] and Au [7]. More recently, sp-ICPMS has been investigated as a tool for the determination of NPs, both fundamentally [8], as well as for various applications: Au in bio-analytical samples [9, 10], Pb in airborne particles [11], dissolved and particulate Ag [12, 13]. In this technique, the sample, an aqueous suspension, is introduced continuously into an ICPMS system that acquires data with a high time resolution. Before introduction, the sample is diluted to a degree that particles enter the plasma one at a time. Following nebulization, a fraction of the nanoparticles enter the plasma where they are vaporised and the individual atoms are ionised resulting in a cloud of ions. This cloud of ions is sampled by the mass spectrometer and detected as a signal pulse in the detector. A typical run time is 60 s and produces a time scan. Under certain conditions (sufficiently dilute samples, known and fixed composition of the particles etc.), the number of pulses detected per second is directly proportional to the particle number concentration in the sample while the intensity of the signal pulse is directly proportional to the mass of the detected nanoparticle. Assuming a certain particle shape (e.g. spheres) and composition, one can calculate the diameter of the particle. The method has the advantage of being fast, is still number-based and, by virtue of the mass spectrometric detection, very selective. Naturally, its applicability is limited to particles of well-defined composition, as otherwise no relation between size and signal exists. The detection power of the method is limited by the presence of ionic analytes of the same material. The higher the dissolved background, the larger particles must be to be reliably distinguishable from the background. Therefore, particles consisting of rather rare elements like Ag have a lower size limit of detection than particles consisting of common compounds like silica. Preliminary data suggest that the size limit of detection for AgNPs is about 20 nm.
Like most other methods used for the sizing of nanoparticles, it also cannot distinguish between single and constituent particles. Nevertheless, if found accurate enough, sp-ICPMS therefore could be a useful method for the implementation of the EC definition as long as the particles can be dispersed into their constituent particles.
Shrink films, cutting boards and storage boxes containing AgNPs are offered on the internet (http://factory.dhgate.com/metallized-film/nano-silver-antimicrobial-cling-film-bag-p47593893.html, http://www.gaswatch.com/products/13%22-x-11%22-Antimicrobial-Round-Cutting-Board.html, http://hznano.en.alibaba.com/product/605559706-213764856/Nano_silver_antimicrobial_Storage_Box_manufacturer.html). It is not unthinkable that AgNPs migrate from these materials into foodstuffs. Therefore, AgNPs were selected as target analyte for the validation of sp-ICPMS. This paper describes the first method validation study for sp-ICPMS for the sizing and quantification of AgNPs in aqueous media.
Experimental
Study concept
The main goal of the study was the evaluation of precision and trueness of the size determination using sp-ICPMS covering the size range of particle diameters from 20 to 100 nm. Also the precision of the particle number concentration was evaluated but was deemed of less importance as current legislation does not put any limits on absolute NP concentrations. The lower size limit of 20 nm (rather than the 1 nm stipulated by the EC definition) was selected as preliminary results of the in-house validation indicated a limit of detection around 20 nm, thus rendering use of even smaller particles not meaningful.
Precision data were determined from the agreement of the results from the participants. By comparing results from different methods obtained on the materials, an indication of presence or absence of a systematic bias could be obtained.
All participants used the same standard operating procedure (SOP) but were free in their choice of calibration standards.
Two standard food simulants, namely, de-ionised water and 10 % v/v ethanol, were selected as test matrix for this first intercomparison.
Test samples
Spherical, monodisperse AgNPs of 20-, 40- and 100-nm nominal diameter were obtained from NanoComposix (NanoComposix, San Diego, USA). The particles were from the company’s NanoXact product line (20-nm PVP NanoXact silver; EAW1126; 40-nm PVP NanoXact silver; JME1115; 100-nm PVP NanoXact silver; EAW 1169; all PVP stabilised). A blank sample consisting of de-ionised water coloured with a mixture of yellow and blue food dye obtained from a local supermarket was included in the set to detect false positives. Of each suspension, 1.5 mL was filled at IRMM in 2-mL polypropylene tubes of the brand Axygen “Maximum Recovery” (Axygen Inc, Union City, USA). Thirty vials of each materials were filled. Particle diameters given by the company were largely confirmed by sp-ICPMS and centrifugal liquid sedimentation (CLS), respectively, although differences for the largest particles were noted. Properties of the samples are given in Table 1.
Homogeneity of the suspensions in the vial was assessed by sp-ICPMS. The 1st, 15th and 30th vial of each materials were tested by sp-ICPMS in two replicates each. The standard deviation of the median particle diameters ranged from 4.2 % (20 nm) to 0.9 % (100 nm). This means that for between-laboratory standard deviations above 14 % (20 nm) to 3 % (100 nm), between-sample variation does not affect the variation between laboratory results. As shown in the results (Section 3.2), this condition is met and the samples therefore fulfil the ISO 13528 [14] requirement for sufficient homogeneity. Concentrations of particulate Ag varied significantly more: relative standard deviations of the six results ranged from 16 to 26 %. However, as will be shown below, this variation is still negligible compared with the standard deviation observed for particle number concentration.
Stability of the suspension in the vials was assessed by UV absorbance and by centrifugal liquid sedimentation (CLS): Once per week, the UV absorption maximum (which correlates with the size of AgNPs) was measured and the particle-size distribution was determined by CLS according to ISO 13318-1 [15] using a working instruction validated for silica [16]. In addition, a second set of sp-ICPMS measurements was performed after receiving the results. These tests showed that the median particle diameter and total Ag mass fraction remained unchanged for all three materials for the complete duration of the study. The sp-ICPMS data indicated a decrease in the particle number concentrations over these 3 months by 40 to 60 %. Converting this maximum degradation of 60 % into a standard uncertainty using a rectangular distribution results in an uncertainty of stability of 17 %, which, using the criteria of ISO 13528 [14], is negligible compared with the between-laboratory variation eventually found in this intercomparison (see Section 3.2.2). It should be pointed out that this potential decrease of the number concentration is contradicted by the participants’ results, which do not show any trend with time (data not shown).
Therefore, the homogeneity and stability studies demonstrated that homogeneity and stability would not affect the between-laboratory variation for the median particle diameter, the main target of the study. Homogeneity and stability were worse for the particle number concentration, but also negligible compared with the variation between laboratories in this intercomparison.
Participants
Twenty-three laboratories from academia, regulators as well as industry were recruited by screening the literature as well as by direct invitation via contact lists of current EU projects and notification via the ISO technical committee on nanotechnologies (ISO/TC229). Thirteen of them had prior experience with sp-ICPMS, albeit in several cases only for a short time or were in the process of establishing the methodology in their laboratories. The other laboratories had prior experience in the handling of NPs and ICPMS, but not of the combination or only ICPMS experience.
Nineteen of the laboratories were European, and two laboratories each came from the USA and Canada.
Measurement procedure
A detailed standard operating procedure (SOP) was sent to all participants. In brief, the method consists of setting the ICPMS dwell time to 3 ms with a total acquisition time of 60 s giving 20,000 separate bins. If the samples are highly diluted, the presence of two particles in the plasma at the same time is unlikely. Using data from a Poisson distribution reveals that if on average 2,000 particles are detected over this 20,000 bins, the fraction of events arising from doublets/triplets is below 5 %. A simulation using MS Excel shows that these multiplets introduce a bias of <0.15 nm for 20- and 40-nm particles, which is negligible.
Ionic Ag standards in aqueous solution with concentrations between 0.2 and 5 μg/L were prepared. Calibration with these standards allows calculation of the number of atoms in each particle. The nebulisation efficiency, a prerequisite for the correct mass and particle number quantification, is determined using Au nanoparticles of known concentrations. The samples are diluted to fall within the calibration range of the method. This analytical method was validated at RIKILT according to 2002/657/EC, and performance characteristics are in accordance with this Commission Decision.
Currently, no commercial software for the evaluation of sp-ICPMS is available. Therefore, a dedicated Excel-spreadsheet was developed and provided to all participants.
A dedicated training workshop was held to make the participants familiar with the procedure. This training workshop was followed by a telephone conference for those participants that could not participate in the workshop.
Study protocol
Preliminary tests had shown that AgNPs in the diluted test matrices were not stable. Therefore, participants were required to prepare test preparations by spiking de-ionised water or 10 % ethanol with one of the four materials each.
Participants received one vial of each of the four materials together with a temperature datalogger to demonstrate the absence of excessively high or low temperatures during transport. The following eight preparations had to be prepared:
-
Four preparations of AgNPs (of which one blank) in de-ionised water by adding 0.25 mL of the materials into 49.75-mL de-ionised water.
-
Preparations of AgNPs (of which one blank) in 10 % ethanol water by adding 0.25 mL of the materials into 49.75-mL 10 % ethanol.
Each preparation contained only one material.
Each participant was asked to measure each of the eight preparations three times and to report median particle diameter, Ag mass concentration and particle number concentration.
The results were reported on a spreadsheet that was returned both electronically as well as asigned hardcopy.
Data evaluation
Evaluation of the results was performed according to ISO 5725-2 “Accuracy (trueness and precision) of measurement method and results” [17]. Results were screened using the graphical consistency technique (Mandel’s h and k-plots) to test for consistent bias or high variances, and laboratories with consistently high bias or variances were excluded. The remaining datasets were tested for outlying means and variances using the Grubbs and Cochran procedure. Outliers at a 99 % significance level were removed from the dataset. The remaining datasets were tested for normality using normal probability plots, and skewness and kurtosis tests. Repeatability standard deviation (s r), standard deviation between laboratories (s L) and reproducibility standard deviation (s R) were calculated from the mean squares within group (MSW) and mean squares between groups (MSB) using one-way analysis of variance (ANOVA):
Finally, it was checked whether there is a relationship between particle diameter and these statistical parameters for the method. When no such relationship existed, a summary standard deviation \( \overline{s} \) was calculated from six standard deviations s i of the six preparations actually containing AgNPs:
If such a relationship existed, it was checked whether the relative standard deviations would be constant. If yes, average standard deviations were calculated according to Eq. 4, using the relative standard deviations instead.
The data evaluation was performed using the characterisation module of the Program SoftCRM [18].
Results and discussion
Twenty-one of the 23 participants submitted results. The remaining two laboratories were hampered by software problems—their ICPMS software did not allow setting a dwell time small enough for the method.
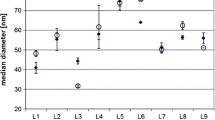
Results for median particle diameter, Ag mass concentration and Ag particle concentration for the 40-nm particles in water are shown in Fig. 1. The data for all results are given as electronic supplementary material.

Results for median particle diameter, Ag mass fraction and Ag particle number concentration in preparation 3 (40-nm AgNPs in de-ionised water)
Two laboratories did not report results for all preparations. In addition, two laboratories reported results for the 20-nm materials as “<LOD”. These results were not used for the numerical evaluation.
Selectivity
Two laboratories reported particles in the two blank preparations although the material for the two blanks had been tested and found to be Ag-free. One of the two laboratories reported particle number concentrations four orders of magnitude below the results of the other preparations, demonstrating that this finding of particles is an analytical artefact. The other laboratory reported particle number concentrations for the blank that were about 1/10 of its findings for the other suspensions, thus constituting a real false positive.
The results show that, as expected, sp-ICPMS is, by virtue of the highly selective MS, selective. However, as with any method, particle diameter information should be taken in combination with particle number information to prevent false positives.
Precision
Median particle diameter
Mandel’s h plot revealed that one laboratory had submitted consistently high results. All data from this laboratory were therefore eliminated.
After elimination of outliers, results for preparation 1 are Gaussian-distributed, but follow a very broad distribution. Results range from 12.7 to 65 nm with an average median diameter of 41.5 nm. Results for preparation 3 seem to follow a trimodal distribution with populations around 30, 40 and 60 nm. However, the skewness and kurtosis tests indicate normality. Preparation 4 follows a broad Gaussian distribution with results ranging from 55 to 111 nm.
The results of the 10 % ethanol preparations show a similar picture: data for preparation 5 follow closely a (very broad) Gaussian distribution, with data ranging from 10 to 61 nm. Similar to preparation 3, data for preparation 7 show two clusters, one containing results from 31 to 47 nm, the other four results between 57 and 65 nm, but again skewness and kurtosis tests indicate normality. A similar picture is visible for preparation 8 with a cluster of six results between 36 and 65 nm and one of 11 results between 80 and 102 nm. Also here, normal probability plot and skewness/kurtosis tests do not allow discarding the assumption of normality.
Number of datasets, number of outliers, mean, s r, s L and s R are shown in Table 2.
Repeatability standard deviations were very low for all preparations, ranging from 0.6 to 1.7 nm. Contrary to this, between-laboratory standard deviations are rather high, ranging from 12 to 20 nm. As those similar averages were obtained for the 20- and 40-nm suspensions, one might argue that the limit of detection is 40 nm. In fact, two laboratories reported results as <22 and <40 nm for suspension 1. A closer inspection of the data shows that the limit of detection is highly laboratory-dependent: out of the 15 laboratories that submitted results for all three particles in at least one medium, six could clearly distinguish the 20-nm particles from the 40-nm particles, demonstrating that 20 nm is clearly above their limit of detection, whereas the other nine laboratories measured the same size for 20- and 40-nm particles. However, sizes measured for the 20-nm particles among the six laboratories still vary between 10 and 42 nm, indicating considerable proportional and constant bias. Results within each laboratory are consistent at higher sizes, allowing clear distinction between the 40- and 100-nm particles: a Youden graph (Fig. 2), plotting the results for preparations 3 and 7 versus preparations 4 and 8 for each laboratory, shows two distinct populations, both of which lie closely to a straight line. This indicates that most laboratories show a proportional bias compared with the other laboratories (linear relationship) and that at least one population shows a constant shift for preparation 3/7 (shift between the two populations). This constant difference is about 20 nm, exactly the average difference between the target value for preparations 1 and 3. This indicates that further training, combined with improvements in calibration, could significantly reduce the method variability.

Youden plot of results for preparations 3 and 4 versus results for preparations 7 and 8. The lines show approximately the shift separating the two populations
Restricting the laboratories to those laboratories that had prior experience with nanoparticle analysis and ICPMS (although not necessarily with sp-ICPMS) gives only marginally improved results: repeatability standard deviation range from 0.6 to 1.8 nm and reproducibility standard deviations range from 8.5 to 20.5 nm.
Precision was not correlated with particle diameter. As there is no significant influence of the particle size on the standard deviations, average within repeatability, between-laboratory and reproducibility standard deviations can be calculated as \( {\overline{s}}_r \) = 1.0 nm, \( {\overline{s}}_L \) = 14 nm and \( {\overline{s}}_R \) = 14 nm, respectively.
Relative repeatability standard deviations range from 1.3 to 4 %, whereas reproducibility standard deviations range from 20 to 35 % (the lowest always being for the highest particle size). This is significantly worse than the results obtained in an intercomparison for dynamic light scattering (DLS) and CLS, where within-laboratory standard deviations of 0.6 to 1.4 % and between-laboratory standard deviations of 5–6 % were achieved [19]. The results in this intercomparison therefore are significantly worse. In our view, several factors contribute to the currently better performance of DLS and CLS: both methods are long-established and dedicated instruments and ISO standards are available. Participants in the intercomparison described in [19] were carefully selected for experience with the methods. Contrary to this situation, few participants in this study had thorough prior experience with sp-ICPMS. Although a training workshop had been organised, questions asked during the study indicated room for improved familiarity with the method. Increased experience is expected to result in better reproducible results. This means that while sp-ICPMS can currently not match long-established methods in precision, it shows promise as a screening technique, especially taking into consideration that it delivers number-based particle distributions. If the method is improved, it might even develop from a screening method into a confirmatory method.
Particle number concentration
After elimination of outliers, results for preparation 1 are Gaussian-distributed. Removal of the two outliers in preparation 3 yielded a bimodal dataset with one cluster of results from 0 to 50 billion particles per litre and the second cluster from 100 to 220 billion particles per litre. Several more outliers were found for preparation 4. As this hinted at “snowballing”, the elimination of outliers was stopped. The data followed a normal distribution. Two Grubbs and one Cochran outlier were removed from the data for preparation 5, yielding a normal distributed dataset. Four outliers were removed from the dataset for preparation 7. The remaining data formed two clearly distinct clusters, one with results from 0 to 50 billion particles per litre and one with 150 to 220 billion particles per litre. For preparation 8, a Gaussian-distributed dataset was obtained after removal of one Grubbs and three Cochran outliers.
The determination of the particle number concentration produced many more outliers than the median particle diameter. The reason for this high number of outliers is not clear. One contributing factor could be that some participants reported particle number concentrations as determined (i.e. in the diluted preparation), some in the preparation and some in the original vial. To exclude this effect, participants were asked repeatedly to confirm the basis of their calculation. A second contributing factor could be loss of particles over time, as indicated by the stability study. However, the magnitude of outliers (sometimes several orders of magnitude) makes this explanation unlikely and the question remains open. Although there were significantly more outliers, no laboratory showed consistently biassed results of consistently high standard deviations.
The number of retained datasets, the mean of laboratory means and the results of the one-way ANOVA for s r, s L and s R are listed in Table 3.
As was the case for particle diameter, repeatability standard deviations are significantly smaller than between-laboratory standard deviations. The bimodal distributions of the data from preparations 3 and 7 indicate that these data should not be used to calculate a correlation between particle number concentration and precision. This leaves only four results to draw up the regression line. With so few results, any test for significance is bound to be negative (critical t-value for 2 degrees of freedom and 95 % confidence interval is 4.3). As repeatability and reproducibility standard deviations seem to increase with the particle number concentration (Table 3), relative s r, s L and s R (excluding the data from preparations 3 and 7) are roughly constant and calculated as \( {\overline{s}}_r \) = 11 %, \( {\overline{s}}_L \) = 78 % and \( {\overline{s}}_R \) = 79 %.
Especially the reproducibility standard deviation is very high. However, when assessing this figure, one must take into consideration the low number of particles measured, which is significantly below analyte concentrations in water analysis. Interlaboratory comparisons for trace organics in water yielded reproducibility standard deviations of 32 % for methyl-tert-butylether in water at a concentration of 5 ∗ 1014 molecules/L [20] and of 15–20 % for 2,4-D at the same concentration level [21]. What is more, relative reproducibility standard deviations of 100 % and more have been reported in the early days of mycotoxin analysis [22], highlighting the problems in any new field of analysis. Then higher standard deviation between laboratories for sp-ICPMS may therefore be expected from the relative novelty of the method.
Trueness
Median particle diameter
Evaluation of trueness is difficult, as particle diameter is a method-defined measurand. This means that diameters obtained by different methods are generally not comparable, unless the particles are mono-modal and spherical. For the particles used, these conditions are sufficiently met to make the comparison across methods meaningful. The median particle diameters as determined in the intercomparison are compared with the results by TEM (provided by the supplier of the particles) and CLS in Table 4.
The results of the 20-nm suspension are higher than those obtained by TEM and CLS. One reason for this high finding could be that sp-ICPMS is here close to its size limit of detection. This is confounded by two participants reporting results as “<LOD” for the smallest particles. The bias for the 100-nm particles could be explained by the indication of the bimodality for the results for preparations of suspension 5, a bias in the TEM determinations performed by the supplier of the materials. Regardless of these biases, results for the particles with nominal 40 and 100 nm are by and large in line with the target values.
There was no significant difference between results obtained in the aqueous or ethanol-based preparations. This indicates that the method is robust against such changes in matrix. This finding is not unexpected as potential matrix effects are largely diluted away. Although the uncertainty is high, this result is a positive sign for the capability of the method.
Particle number concentration
No definite value for the particle concentration in the original suspension is available. Information from the producer was judged the most reliable, albeit imperfect surrogate for a target value. The mean particle number concentrations in the undiluted suspensions as determined in the intercomparison are compared with the specifications of the supplier in Table 5.
As was the case for the Ag mass concentrations, particle number concentrations are significantly below those specified by the producer. This difference was especially marked for the 20-nm material, most likely caused by many particles being below the size limit of detection and thus going undetected. The lower finding for the other two materials might indicate dissolution and therefore vanishing of particles, but could also indicate overlooking a significant fraction of particles. The data available do not allow a clear decision between these two possibilities.
As with the particle diameter determination, there was no significant difference between the results obtained in the aqueous and ethanol-based preparations. This indicates that the method is robust against such changes in matrix. This finding is not unexpected as potential matrix effects are largely diluted away. Although the uncertainty is high, this result is a positive sign for the capability of the method.
Conclusions
Repeatability, between-laboratory and reproducibility standard deviations for particle diameter are 1 nm (s r) and 14 nm (s L and s R). While repeatability is good, reproducibility is more than one order of magnitude above repeatability, showing ample need and opportunity for improvement in comparability among laboratories. Precision is still significantly worse than that obtained by long-established and standardised methods like DLS or CLS. Further training as well as better calibration could be expected to improve agreement among laboratories. The average median particle diameter as determined by sp-ICPMS differs at the upper and lower range of the investigated interval from results measured by TEM and CLS, with apparent positive bias at low and negative bias at high diameters. However, the deviation around the critical value of 100 nm is small enough to allow screening for the presence of nanoparticles. Two participants reported results as “<20 nm” and “<40 nm”, indicating that the size limit of detection is in this range. The study showed that identification of blanks is possible if information on the particle number concentration or mass concentration of particles is also taken into account. If not, a small number of particles will still give a median diameter.
Relative repeatability and reproducibility standard deviation for Ag particle number concentration were estimated 11 and 78 %, respectively. While being high, these precision data compare well with precision of organic trace analysis when the very low concentrations of particles (several orders of magnitude below molecular concentrations in organic trace analysis) and the novelty of the method are considered. Also here, reproducibility standard deviations differed more than usual from repeatability standard deviations, indicating room for method improvement by better calibration and further training.
Results for Ag particle number concentration are significantly lower than the specifications from the manufacturer. At present, it is unknown whether these findings are due to method bias or due to dissolution of particles.
No differences were found between samples prepared in water or 10 % ethanol, demonstrating robustness of the method regarding the solvent within the scope of the study.
In summary, taking into consideration the novelty of the method, the fact that the vast majority of participants had just started establishing the method in their laboratories and the lack of suitable certified reference materials for calibration, the accuracy of sp-ICPMS, is already satisfactory for screening purposes when limited to particle diameter. Even in its current state, the method, providing number-based results, is, within the scope of the method (particles of defined composition and shape; dispersibility of particles and no distinction between single and constituent particles), a useful tool for determining whether a material fulfils the EC definition of nanomaterial. In its current state, the method can only approximately quantify the amount of nanoparticles (mass or particle number concentration). This has few implications, as there are no current limits for the amount of nanoparticles.
While further improvements of the method, especially with respect to software tools for evaluation, hardware options for shorter dwell times, calibration standards for determining nebuliser efficiency and further experience by laboratories are certainly desirable, the results of this study demonstrate the suitability of sp-ICPMS for the detection and quantification of certain kinds of nanoparticles.
References
Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products
Regulation (EU) No 1169/2011 of the European Parliament and of the council of 25 October 2011 on the provision of food information to consumers
Recommendation 2011/696/EU of the European Commission of 18 October 2011 on the definition of nanomaterial
Linsinger TPJ, Roebben G, Gilliland D, Calzolai L, Rossi F, Gibson N, Klein C (2013) JRC Reference Report ‘Requirements on measurements for the implementation of the European Commission definition of the term ‘nanomaterial’ EUR25404EN,Publications Office of the European Union, Luxembourg, ISBN 978-92–79-25602-8
McCarthy J, Degueldre C (1993) Characterisation of environmental particles, Vol 2, 6th edn. Lewis Publishers, Chelsea MI, pp 247–315
Degueldre C, Favarger PY (2003) Colloid analysis by single particle inductively coupled plasma-mass spectroscopy: a feasibility study. Colloids Surf A 217:137–142
Degueldre C, Favarger PY, Wold S (2006) Gold colloid analysis by inductively coupled plasma-mass spectrometry in a single particle mode. Anal Chim Acta 555:263–268
Pace HE, Rogers NJ, Jarolimek C, Coleman VA, Higgins CP, Ranville JF (2011) Determining transport efficiency for the purpose of counting and sizing nanoparticles via single particle inductively coupled plasma mass spectrometry. Anal Chem 83:9361–9369
Scheffer A, Engelhard C, Sperling M, Buscher W (2008) ICP-MS as a new tool for the determination of gold nanoparticles in bioanalytical applications. Anal Bioanal Chem 390:249–252
Yu L, Andriola A (2010) Quantitative gold nanoparticle analysis methods: a review. Talanta 82:869–875
Suzuki Y, Sato H, Hikida S, Nishiguchi K, Furuta N (2010) Real-time monitoring and determination of Pb in a single airborne nanoparticle. J Anal At Spectrom 25:947–949
Laborda F, Jiménez-Lamana J, Bolea E, Castillo JR (2011) Selective identification, characterization and determination of dissolved silver(I) and silver nanoparticles based on single particle detection by inductively coupled plasma mass spectrometry. J Anal At Spectrom 26:1362–1371
Mitrano DM, Lesher EK, Bednar A, Monserud J, Higgins CP, Ranville JF (2011) Detecting nanoparticulate silver using single-particle inductively coupled plasma-mass spectrometry. Environ Toxicol Chem 31:115–121
ISO 13528:2005 “ Statistical methods for use in proficiency testing by interlaboratory comparisons”, ISO, Geneva, 2005
ISO 13318:2001 “ Determination of particle size distribution by centrifugal liquid sedimentation methods—Part 1: General principles and guidelines”, ISO, Geneva, 2001
Braun A, Couteau O, Franks K, Kestens V, Roebben G, Lamberty A, Linsinger TPJ (2011) Validation of dynamic light scattering and centrifugal liquid sedimentation methods for nanoparticle characterisation. Adv Powder Technol 22:766–770
ISO 5725:1996 “Accuracy (trueness and precision” of measurement method and results, ISO, 3rd revision, Geneva 1996
Bonas G, Zervou M, Papaeoannou T, Lees M (2003) “SoftCRM”: a new software for the certification of reference materials. Accred Qual Assur 8:101–107
Braun A, Kestens V, Franks K, Roebben G, Lamberty A, Linsinger TPJ (2012) A new certified reference material for size analysis of nanoparticles. J Nanoparticle Res 14:1012–1023
Schuhmacher R, Führer M, Kandler W, Stadlmann C, Krska R (2003) Interlaboratory comparison study for the determination of methyl tert-butyl ether in water. Anal Bioanal Chem 377:1140–1147
Kandler W, Schumacher R, Roch S, Schubert-Ullrich P, Krska R (2004) Evaluation of the long-term performance of water-analyzing laboratories. Accred Qual Assur 9:82–89
Horwitz W, Kamps LR, Boyer KW (1980) Quality assurance in the analysis of foods and trace constituents. J Assoc Off Anal Chem 63:1344–1354
Acknowledgments
The authors wish to acknowledge Guy Auclair, Ringo Grombe, Vikram Kestens and Yannic Ramaye for performing the CLS and UV spectrometry measurements for the homogeneity and stability testing of the suspensions and Ana Boix and Gert Roebben (all JRC-IRMM) for the review of the data. We also thank the participants in the study for their good cooperation and openness:
Sebastian Sannac (Agilent, Les Ulis, FR), Paul Westerhoff (Arizona State University, Engineering Center, Tempe, US), René Chemnitzer Bruker (Berlin, DE), Jutta Tentschert (Bundesinstitut für Risikobewertung (BfR), Berlin, DE), Alan J Lawlor (Centre for Ecology and Hydrology, Lancaster, UK), Claus Wiezorek (Chemisches und Veterinäruntersuchungsamt Münsterland-Emscher-Lippe (CVUA-MEL), Münster, DE), James Ranville (Colorado School of Mines, Department of Chemistry/Geochemistry, Golden US), Douglas Gilliland, Claudia Cascio (EC-Joint Research Centre, Institute for Health and Consumer Protection, Ispra, IT), Malcolm Baxter (Food and Environment Research Agency (FERA), York, UK), Francesco Cubadda, Federica Aureli (Istituto Superiore di Sanità, Dept. Food Safety and Veterinary Public Health, Rome, IT), Olf Richter (Landesuntersuchungsamt Sachsen, Fachgebiet 2.7, Dresden, DE), Chady Stephan (PerkinElmer Canada, Woodbridge, CA), Petra Krystek (Philips Innovation Services, Eidhoven, NL in cooperation with KWR, Eindhoven, NL), Zahira Herrera Rivera (RIKILT Wageningen UR, Wageningen, NL), Carl Isaacson (Swiss Federal Institute of Aquatic Science and Technology (EAWAG), Dübendorf, CH), Andrea Ulrich, Sabrina Losert (Swiss Federal Laboratories for Materials Science and Technology, Dübendorf, CH), Erik H Larsen (Technical University of Denmark, Department of Food Chemistry, Søborg DK), Daniel Kutscher (Thermo Fisher Scientific, Bremen,DE), Geert Cornelis (University of Gothenburg, Department of Chemistry and Molecular Biology, Göteborg, SE), Kevin Wilkinson (University of Montreal, Department of Chemistry, Montreal, CA), Frank von der Kammer, Stephan Wagner (University of Vienna, Department of Environmental Geosciences, Vienna, AT), Francisco Laborda (University of Zaragoza, Institute of Environmental Sciences, Zaragoza, ES), Sara Totaro (Veneto Nanotech, ECSIN Laboratories, Rovigo, IT).
The work leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement no 245162.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Characterisation of Nanomaterials in Biological Samples with guest editors Heidi Goenaga-Infante and Erik H. Larsen.
Rights and permissions
About this article
Cite this article
Linsinger, T.P.J., Peters, R. & Weigel, S. International interlaboratory study for sizing and quantification of Ag nanoparticles in food simulants by single-particle ICPMS. Anal Bioanal Chem 406, 3835–3843 (2014). https://doi.org/10.1007/s00216-013-7559-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-7559-9