
Abstract
We describe a capillary electrophoresis-mass spectrometry (CE-MS) method for newborn screening of a representative amino acid metabolic disease, namely, phenylketonuria (PKU). Underivatized phenylalanine and tyrosine in a dried blood spot (DBS) were simultaneously determined by CE-MS equipped with an ionophore membrane-packed sheathless electrospray ionization interface, which was developed by our group. The method was optimized for rapid determination of the underivatized amino acids, phenylalanine and tyrosine extracted from a DBS. Under the optimized conditions, the limit of detection of phenylalanine and tyrosine (signal-to-noise ratio, 3) was 0.03 and 0.07 mg/L in DBS, respectively, with a CE run time of less than 3 min. For repeated runs of a sample, coefficients of variation (CVs) for migration time were less than 3.7 %, whereas CVs for the area ratio under the curve were 2.1 and 2.9 % for 20 consecutive runs of 49.5 mg/kg Phe and 36.2 mg/kg Tyr, respectively. However, the relative standard deviations of intra- and interday assays for DBS samples were <6.2 and <5.8 %, respectively, which were substantially due to sample extraction from DBS. The analytical method was applied to real clinical samples of Korean neonates, and results were compared with those of conventional methods for PKU diagnosis, which required reference analytical methods such as isotope dilution CE-MS or high-performance liquid chromatography-mass spectrometry for quality assurance of the conventional kit-based assays. The distinct advantages of high sensitivity and extremely low sample volume, as well as a simple, easy, and economic sample pretreatment, were demonstrated for the proposed method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The aim of newborn screening is the detection and diagnosis of inherited metabolic diseases (IMDs) in the neonatal period. The majority of IMDs are autosomal recessive genetic disorders that can induce severe irreversible complications through the accumulation of toxic metabolites by congenital metabolic enzyme deficiencies. Early medical intervention in many cases provides the only possibility to avoid physical and mental retardation or even death. These diseases can be diagnosed by determining the accumulated metabolites in the urine, plasma, or blood [1–3]. Amino acid metabolic disorders were one of the earliest IMDs investigated on a large scale. In particular, phenylketonuria (PKU; OMIM 261600) is an autosomal recessive genetic disorder characterized by an enzyme deficiency in phenylalanine (Phe) hydroxylase (OMIM 612349). Among a number of IMDs, PKU is a fairly common disease with a high frequency in Caucasian (approximately 1:4,500–1:9,000) and Asian (1:110,000 in Japan and 1:55,000 in South Korea) populations. PKU has critical effects on brain development and leads to progressive mental retardation, but it can be avoided if treated soon after birth with a low Phe diet [4–6]. This stimulated the search for a reliable screening method in neonates, and neonatal screening programs have been implemented for neonates at the national level in many countries. Typically, Phe hydroxylase deficiency increases the blood Phe concentration, which has been used as a biomarker for PKU screening. Phe hydroxylase catalyzes the conversion of Phe to tyrosine (Tyr), and the concentration ratio of Phe-to-Tyr (Phe/Tyr) would therefore be elevated in PKU when compared with normal neonates. For this reason, the use of a Phe/Tyr value has also been suggested to reduce false-positive errors [6, 7]. For newborn screening, specially designed filter paper has been used for dried-blood-spot (DBS) sampling. The use of a DBS obtained from a baby by spotting a little blood from a heel or toe prick onto filter paper was first introduced by Dr. Guthrie for the detection of PKU in the early 1960s [8]. This novel approach of human blood collection led to the population screening of newborns and other clinical tests. Nowadays, DBS is applied widely in many other fields including therapeutic drug monitoring, toxicokinetics, and pharmacokinetics. It offers many advantages over conventional whole-blood, plasma, or serum sample collection methods since it utilizes a small sample volume, employs an easy and less-invasive sampling technique, involves simpler handling and storage, and facilitates easier transfer. It has also been reported that the use of a DBS can offer advantages in stabilizing unstable compounds [9–12]. However, some factors limit the applicability and acceptance of DBS quantification from an analytical point of view. Indeed, it has been demonstrated that factors such as hematocrit, blood viscosity, analyte nature, DBS card substrate, paper chromatography effect, spotting technique, and spotting conditions can lead to variations in the dimensions of spots and an uneven distribution of analytes within a spot or between spots [13–16]. Although there is no doubt regarding the effectiveness of DBS, the use of this procedure requires careful consideration of the potential influences of such factors on measurement uncertainties and the interpretation of results.
Some biochemical methods, such as a bacterial inhibition assay (BIA) [17] and enzymatic colorimetry assay (ECA) [18] measure Phe semiquantitatively. These methods are used widely in the form of commercial kits because of simplicity and low cost but have the disadvantages of low accuracy and low sensitivity. Although effective in high-throughput screening in a clinical setting, these kits may lead to relatively high rates of false-positive results. Instrumental methods of high-performance liquid chromatography (HPLC), GC, and capillary electrophoresis (CE) with various detection schemes including mass spectrometry (MS) have also been developed for accurate PKU detection based on their high sensitivity and specificity. Although the initial implementation cost may be high, these methods can serve as a reference method for quality control and quality assurance of screening with commercial kits [3–7, 19].
Hyphenation of capillary electrophoresis with mass spectrometry (CE-MS) has emerged as a powerful analytical approach as it is particularly well suited for the analysis of complex biological samples of limited quantities. Although the on-line interfacing of CE with MS is more problematic than the interfacing of HPLC with MS, the combination of high-resolution CE separation and high selectivity of MS detection is sufficiently attractive for use in various fields. Consequently, a number of methods to interface CE and MS have been devised [20–22]. Due to its easy implementation and compatibility with MS connection, commercially available CE-MS instruments have favored to adopt a sheath-flow interface. More recently, however, sheathless interfaces have gained popularity as researchers seek to improve detection limits. Reported sheathless interface strategies introduced a sharpened or tapered and coated capillary tip-end for electrical contact, as well as two capillaries butted together via interfaces designed from microdialysis tubing, stainless steel tubing, micro-tee, etc. with time-consuming fabrication procedure. For widespread applications of CE-MS, particularly with electrospray ionization (ESI), the stability of operation and durability for long-term uses are essential. Easy fabrication and quick replacement would also be advantageous in routine applications. In this regard, we reported previously that our two-capillary sheathless CE/ESI-MS interface with an ionophore membrane-packed electro-conduction channel satisfied such requirements, and that creatinine/uric acid [22] and underivatized amino acids [23] were successfully analyzed. Simplicity and ease of fabrication and replacement of capillary tubing were also advantageous. More importantly, the interface operated stably and can last long enough for repeated analysis without any hydro-organic additives in BGE. In this work, we tested the feasibility of the sheathless CE-ESI/MS interface in the form of routine application. As described above, simultaneous determination of underivatized Phe and Tyr extracted from clinical DBS specimens was attempted for PKU diagnosis. We were also interested in the possible inhomogeneity issue of DBS sampling and tried to minimize such variation through isotopic-dilution mass spectrometry (ID-MS). The results of the proposed method were compared with those of other conventional methods for PKU diagnosis.
Experimental
Materials
l-phenylalanine (Phe), l-tyrosine (Tyr), isotope-labeled l-phenylalanine (13C9, 95 %; 15N, 95 %; Phe*), isotope-labeled l-tyrosine (13C6, 99 %; Tyr*), ammonium acetate (NH4Ac), ammonium hydroxide (NH4OH) and sodium hydroxide (NaOH) were obtained from Sigma-Aldrich (St. Louis, MO, USA). The stock solutions of Phe, Tyr, Phe*, and Tyr* used for calibration and quantification were prepared gravimetrically with a concentration of 2 g/kg in deionized water (DW) and were stored at −20 °C prior to use. Other reagents and solvents used were all of an analytical grade. DW used to prepare the standard solutions, sample solutions, and background electrolytes (BGEs) was purified using a Millipore Alpha-Q water purification system (Millipore, Billerica, MA), and solvents were degassed and filtered through a membrane filter (pore size, 0.2 μm).
Fused-silica capillaries with uncoated inner walls used in the separation capillary and emitter tip (50-μm i.d. and 365-μm o.d.) were obtained from Polymicro Technologies (Phoenix, AZ). Emitter tips were prepared by grinding one end of a capillary into a conical shape on 1,500-grit sandpaper (CAMI designation) attached to a rotating disk. Nafion® tubing (TT-030; 0.025-in. i.d. and 0.035-in. o.d.) was purchased from Perma Pure (Toms River, NJ). The main body of the interface was made of polycarbonate in a local machine shop (Sysco, Daejeon, South Korea).
Dried blood spots for calibrator
Two different PKU screening kits were purchased, one from the Bio-Rad Diagnostics Group (Quantase™ Neonatal phenylalanine screening) and another from PerkinElmer (Neonatal Phenylalanine kit from Wallac). Each kit has a DBS calibrator for Phe, and the concentrations of Phe in each blood spot were 15.6, 38.2, 73.4, and 151.5 mg/L in the Bio-Rad kit, and 2.6, 19, 32, 56, 102, and 150 mg/L in the PerkinElmer kit. Another DBS calibrator for PKU screening was kindly provided from Sapporo Immuno Diagnostic Laboratory (Sapporo, Japan), and the concentration of Phe in each blood spot was 2, 17, 32, 57, 89, 131, and 182 mg/L.
A set of DBS calibrators for the simultaneous determination of Phe and Tyr was prepared in our laboratory. For preparation of the in-house DBS calibrator, whole blood was collected in EDTA vials from a healthy male adult. Stock solutions containing different concentrations of Phe and Tyr were gravimetrically prepared. The final known concentrations in blood were 0, 8.3, 24.8, 49.5, and 115.5 mg/kg for Phe and 0, 5.4, 18.1, 36.2 and 72.4 mg/kg for Tyr with exception of endogenous contents. Fifty microliters of Phe- and Tyr-enriched blood pools were dispensed onto a filter paper with printed circles 12 mm in diameter (S&S 903 type; protein saver card, lot No. W102; Whatman, Sanford, ME, USA). The DBSs were marked as J0, J1, J2, J3, and J4, and added concentrations of Phe and Tyr were as follows: J0, 0 mg/kg Phe and Tyr; J1, 8.3 mg/kg Phe; 5.4 mg/kg Tyr; J2, 24.8 mg/kg Phe and 18.1 mg/kg Tyr; J3, 49.5 mg/kg Phe and 36.2 mg/kg Tyr; and J4, 115.5 mg/kg Phe and 72.4 mg/kg for Tyr. The DBS calibrators were dried overnight on a dry rack at room temperature and stored in a zippered plastic bag with desiccant at 4 °C.
Clinical samples
DBS samples collected from Korean neonates within 7 days of birth (107 PKU-negative samples and 14 PKU-positive samples) were kindly provided by the Seoul Medical Science Institute (Seoul, South Korea), a national depository of samples obtained from general hospitals. All of the samples were primarily screened using BIA [17]. DBS samples were sealed in a vinyl pack with desiccant and stored at 4 °C before analysis.
Sample preparation
A disc of 3.2 mm (0.125 in.) in diameter was punched out of the 12-mm-diameter spotted DBS with a manual paper punch (Whatman, lot No. 9238030) and placed into a 96-well plate. One hundred microliters of extraction solvent (DW) fortified with Phe* and Tyr* (as internal standards) was added into the well. The concentrations of Phe* and Tyr* were 1.3 and 1.4 mg/kg, respectively. The extraction was carried out by gentle agitation of the 96-well plate on a shaker for 20 min. For sample deproteinization, the extracts were transferred to centrifugal ultrafiltration units with a molecular weight cutoff of 10,000 Da (Nanosep® Centrifugal devices; Pall Corporation, Port Washington, NY), and then centrifuged for 10 min at 12,000 rpm. The filtrates were injected into the CE-MS system.
CE-MS
The sheathless CE/ESI-MS interface was constructed in accordance with previous studies [22, 23]. In the interface body, a 400-μm-diameter “capillary channel” was prepared, and two capillaries (separation capillary and emitter tip) were placed in this channel. The ends of the capillaries were finished prior to joining in the interface body using 1,500-grit sandpaper to avoid the production of debris when butted together. The capillaries were fastened with finger-tightened fittings for standard capillary tubing (F-1245; Upchurch Scientific, Oak Harbor, WA), which facilitated easy replacement of capillaries. An electroconduction channel packed with Nafion was prepared for the region where the capillaries adjoined. A piece of platinum (Pt) wire was wrapped around the interface body as an electrode, and a piece of sponge soaked with an electrolyte was then placed between the conduction channel and Pt wire to provide electrical contacts. The diagram of interface body and CE-MS system and the photos of fabrication process are shown in Electronic supplementary material (ESM; see Fig. S1 and S2). The CE-MS interface described above was installed on a commercial nanospray mounting system (PV-300, PicoView®; New Objective, Woburn, MA). A home-built CE system with two independent bipolar high-voltage supplies was interfaced with a triple-quadrupole mass spectrometer (Quattro Ultima, Micromass, Manchester, UK).
Employment of the sheathless CE-MS system was performed using uncoated fused-silica capillaries with a 50-μm i.d. and a 40-cm separation capillary and a 4-cm emitter tip. Although the emitter tip-end of the MS side had been sharpened with sandpaper, the inner-channel still maintained a diameter of 50 μm to the very end of the tip. The BGE used comprised 3 mM NH4Ac, adjusted to a pH of 10.7 with NH4OH; 25 kV was applied for CE separation, where 2.4 kV was applied to the interface for positive-mode ESI. Samples were electrokinetically injected at 25 kV for 3 s. Phe and Tyr were measured in multiple reaction monitoring (MRM) mode, and the monitored precursors to product ion transitions were as follows: Phe, 165.88 > 119.96; Phe*, 175.95 > 129.10; Tyr, 181.95 > 136.04; and Tyr*, 187.93 > 141.98. Source ionization and fragmentation parameters were optimized prior to sample analysis. Peaks of unlabeled and isotopically labeled compounds were integrated, and the ratio of these two areas was calculated.
Extraction efficiency test
The 3.2-mm diameter pre-cut DBS (PCDBS) [24] and blood mixture solution (16.5 mg/kg Phe and 18.1 mg/kg Tyr) were prepared for the extraction efficiency test. Three microliters of the blood mixture was absorbed onto the PCDBS. In parallel, the same volume of blood mixture solution (3 μL) was also placed in another well and used as a reference of extraction recovery from PCDBS samples. The results were compared with the blood solution (non-absorbed) and DBS (absorbed and extracted from paper). The sample procedure was the same, but the extraction solvent and time were changed to optimize extraction conditions for Phe and Tyr in DBS. DW, methanol, acetonitrile, ammonium bicarbonate (pH 7), 0.1 mol/L of hydrochloric acid (HCl) and 0.1 % (v/v) trifluoroacetic acid were compared as extraction solvents. Each extraction solvent (97 μL) and IS solution (16.5 mg/kg Phe* and 18.1 mg/kg Tyr* in DW, 3 μL) was added to each well (PCDBS or 3 μL blood).
Methods of spiking of an internal standard solution
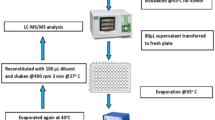
Figure 1 summarizes the preparation procedure concerning addition of the internal standard (ISTD) solution. ISTD was added using several different methods: (a) addition into the extraction solution (control), (b) addition into the blood mixture prior to paper spotting (pre-mixed), (c) ISTD solution spotted solely prior to blood spotting (pre-layered), and (d) ISTD solution layered over a blood spot (post-layered). A green edible dye was added to the IS solution to check its even distribution on the paper for cases (c) and (d). From each blood spot, seven discs were punched out and analyzed to determine any improvement in the analytical precision of DBS.

Sample preparations for the comparative study of ISTD spiking methods in dried blood spots: (a) addition of ISTD into the extraction solution as a control, (b) ISTD addition into the blood mixture prior to paper spotting, (c) ISTD solution spotting prior to blood mixture spotting, and (d) ISTD solution layering over the dried blood spot. Blood mixture the mixture of blood with standard solution, spotting absorbed 50 μL solution onto paper, dry drying overnight on a dry rack at room temperature, ISTD internal standard solution of isotope labeled Phe and Tyr, punched disc 3.2-mm diameter disc from DBS by paper punch, DW distilled water as extraction solvent, extraction 20 min on a shaker at room temperature, ultrafiltration filtration with 10 kD molecular weight cut-off membrane
Method validation
The linearity of Phe and Tyr in DBS were determined using an in-house calibrator in a sample matrix (S&S 903 type; Protein saver card). The intraday and interday precision of the assay were determined from the results of three independent experiments over three consecutive days. Test samples were J0, J1, J2, J3, and J4. After the sample preparation process, all test samples contained 0.8 mg/kg Phe* and 0.9 mg/kg Tyr* as ISTD for ID-MS. The measured value represents peak area ratio of analyte to ISTD. Among the calibrators, J0 was the blank sample that defined the endogenous Phe and Tyr in the blood matrix. After peak integration process, the results for calibration curves were calculated by subtraction of J0 value (measured peak area ratio) from measured J1–4 value, e.g., J1′ = (peak area ratio of J1) – (peak area ratio of J0). The calibration curves were generated with “calculated” values (J1′–J4′) as the y values and the added concentrations of J1–J4 as the x-axis. After that, measured peak area ratio of J0 was also substituted into the calibration curves to determine endogenous concentration of Phe and Tyr in DBS.
The limit of quantification (LOQ) and limit of detection (LOD) of Phe and Tyr of developed CE-MS method were determined by serial dilution of DBS extraction solution (J0) up to signal-to-noise ratios of 3 (LOD) and 10 (LOQ). The LOQ and LOD represented the concentration of Phe and Tyr in the sample solution because one punched disc (3.2 mm in diameter, approximately 3 μL of blood) of DBS for one analysis was diluted approximately 33 times (total volume of sample, 100 μL) before the CE-MS analysis.
Analysis of clinical application and method comparisons
The clinical samples were analyzed by the optimized CE-MS method. The results were compared with the results of a high-performance liquid chromatography-mass spectrometry (HPLC-MS) method and two different ECA methods. The HPLC-MS conditions were optimized based on the analytical conditions published by Jeong et al. [25]. The ECA methods, which utilized Quantase™ Neonatal Phenylalanine Screening (Bio-Rad) and the Neonatal Phenylalanine kit (PerkinElmer), were run in accordance with manufacturer instructions.
Results and discussions
CE-MS conditions
We previously developed a robust sheathless CE-MS interface that worked satisfactorily with an alkaline buffer for the analysis of 12 underivatized serum amino acids [23]. In this work, the conditions used previously were modified for rapid determination of Phe and Tyr. Amino acids are negatively charged at a pH above their pK a2. Although the pK a2 of Phe (9.13) and Tyr (9.11) is similar, Tyr has an acidic side chain with a pK a of 10.07. Therefore, the charge statuses of the two amino acids become different at a BGE pH higher than 10.07, which will facilitate better CE separation. The use of a high BGE pH is also advantageous for stable operation of the two-capillary sheathless ESI interface due to the strong EOF [23]. It was confirmed that a sufficiently fast flow across the emitter tip was essential to maintain a stable electrospray with a sheathless interface. A strong EOF also facilitated rapid detection of both Phe and Tyr (within 3 min). Although the CE separation may not be adequate to reject many potentially interfering materials in complex blood matrices, the extreme high selectivity of MS detection in MRM mode could provide satisfactory specificity. The concentration of the BGE was set to low (∼3 mM) for the stability of ESI, which also contributed to an even faster EOF. The potential problem due to the poor buffer capacity was avoided by frequent replacement of the BGE (replacement after every five runs). Figure 2a shows typical total ion electropherograms of a standard mixture (A-a) and DBS sample (A-b). In Fig. 2 b, typical MRM-electropherograms of Phe, Tyr, and ISTD (Phe* and Tyr*) of a standard mixture are shown. No significant differences were observed from the results of DBS extracts. For repeated runs of a sample, coefficients of variation (CVs) for migration time were less than 3.7 %, whereas CVs for the area ratio under the curve were 2.1 and 2.9 % for 20 consecutive runs of 49.5 mg/kg Phe and 36.2 mg/kg Tyr, respectively. LODs were 0.03 and 0.07 mg/kg for Phe and Tyr, respectively, whereas LOQs were 0.10 and 0.25 mg/kg for Phe and Tyr, respectively.

Total ion electropherograms of Phe and Tyr. Standard mixture (A-a), extracts from dried blood spots (A-b), and MRM channel scan (B). Sample concentrations of standard mixture are 1.65 mg/kg of Phe and 1.81 mg/kg of Tyr and a DBS calibrator with 8.3 mg/kg of Phe and 5.4 mg/kg of Tyr. CE conditions: capillary, uncoated fused silica (40-cm separation capillary and 4-cm emitter tip × 50-μm id); electrolyte, 3 mM NH4Ac (pH 10.7); separation voltage, 25 kV; injection, 3 s at 25 kV; and ESI voltage, 2.4 kV
DBS extraction efficiency
DBS sampling may lead to unsatisfactory analytical performances due to the different physical attributes of common blood specimens. Extraction efficiency from DBS samples was therefore examined carefully using various experimental parameters. For correct assessment of extraction efficiency, the volume of blood spotted in a specific area should be defined. The quantity of blood in a single punched disc could be estimated from the area ratio between the whole-spot area and punched-disc area where the blood volume spotted was accurately known. To avoid uncertainty concerning blood volume and extraction recovery yield, PCDBS with a known blood volume was prepared. Experimental parameters examined included extraction solvent, time, and extraction conditions. Among the tested extraction conditions such as sonication, heating, and homogenization, no significant difference was observed. Extraction solvents were chosen among those used in referenced studies and the several candidates for DBS analysis and MS availability [3, 5, 9, 26, 27]. Extraction efficiency was scored by the degree of agreement between the analytical results for blood and PCDBS. As shown Fig. 3(a), extraction recoveries were rather similar in all cases, except for acetonitrile where DW and HCl showed the best agreements. Organic solvents or acids are frequently employed for simultaneous sample extraction and deproteinization. In this work, however, such solvents caused instability during electrophoresis upon sample injection due to large differences in the composition of the BGE. If sample pretreatment such as evaporation or neutralization was not performed, abnormal peaks resulted. To avoid such complexities, DW extraction followed by ultrafiltration for deproteinization was chosen. In regard to the extraction time, the extraction recoveries of Phe and Tyr from DBS were increased for 10 min, and maintained after that point (Fig. 3(b)). A DBS extraction time of 20 min was finally chosen in this study.

Extraction efficiency tests for Phe and Tyr using PCDBS and the same volume of blood according to the type of extraction solvent (A) and extraction time (B). Blood 3 μL blood mixture solution without paper, DBS PCDBS sample with absorption of 3 μL blood mixture solution, DW distilled water, Me 100 % methanol, ACN 50 % (v/v) acetonitrile, HCl 0.1 M hydrochloric acid, ABC 50 mM ammonium bicarbonate, TFA 0.1 % (v/v) trufluoroacetic acid
Application of an ISTD to DBS sampling
Isotope dilution-mass spectrometry is the most effective way to reduce uncertainties throughout the whole experimental procedure since the isotopic analogues exhibit the same chemical properties as those of unlabeled compounds, except for molecular mass. For this reason, addition of isotope analogues to samples at the beginning of sample preparation will improve analytical traceability and precision. DBS has different physical attributes from other blood samples, and these properties will affect analytical variation. However, ISTD is usually only included in the extraction solution, which does not track the process of solubilizing and eluting the analyte from the DBS to the solution phase. Variations (CVs) of analytical results reported in other studies were up to 6 % [1, 16]. As shown in Fig. 4, variations in our study were 3.40 and 3.19 % for Phe and Tyr, respectively (Fig. 4(a) and (B-a)) when adding ISTD to the extraction solvent. This result represented not only a limit of partial ID-MS but also the importance of full ID-MS to trace the whole procedure including blood sampling and extraction of analyte from DBS.

Sample homogeneity test within a spot (A) and the result of comparisons of CV within a spot with respect to internal standard spiking methods for the preparation of the DBS calibrator (B). y-axis of graph (A) represents peak area ratio of analyte/ISTD, and y-axis of graph (B) represents peak area ratio variations (CV %) of seven discs within one spot
As described in Fig. 1, ISTD can be added at different steps of the analytical procedure. Perhaps the best approach is to mix ISTD with blood samples before spotting (b). However, this may not be practical for a very limited amount of blood obtained by heel or toe pricks. ISTD can be layered onto a DBS before (c) or after blood spotting (d). The impact of the addition of ISTD using the common method (a) was compared with that of three alternative application methods (b–d). As shown in Fig. 4(b), DBS with a pre-mixed blood mixture (b) showed better agreement and a smaller CV than the common method (control (a)). Unfortunately, however, the other two DBSs representing pre- and post-layered forms showed substantially increased variations. In the pre-layered form, a viscosity differential between the ISTD solution (DW) and blood might have worsened the situation. In the post-layered form, it is likely that the physically transformed paper with blood interfered even with the disruption of ISTD. Although this analysis merely investigated a technical issue concerning application of ID-MS for DBS, it clearly demonstrated that the method of ISTD addition in regard to full ID-MS should be viewed in terms of metrological standardization.
Discrepancy among different DBS calibrators
Comparison of three commercial DBS calibrators and an in-house calibrator revealed a substantial discrepancy (see Fig. S3 in the ESM; the calibration curves of Phe in various DBS calibrators for PKU screening). Disagreement was observed not only among different brands but also among the products of the same manufacturer for different batches and different spotted sample volumes. As mentioned earlier, analytical results for DBS samples can vary depending on the manufacturer, type and production lot of paper, as well as the quantity of blood spotted. Moreover, we merely suppose the situation can be worsened by the biological variability resulting from various factors such as different hematocrit contents among babies. Although there have been efforts to develop internationally acceptable DBS reference materials [28, 29], commercial calibrators still show an unacceptable discrepancy. Tuuminen reported that the analytical bias of Phe measurement depends on preparation of the calibrators [14]. Consequently, we used in-house DBS calibrator with known concentrations of Phe and Tyr for simultaneous determination of Phe and Tyr as well as PKU screening.
Method validation
The proposed CE-MS method was first evaluated with our in-house calibrators for four different levels of Phe and Tyr (J1–4). Measurement linearity was also excellent since r 2 values were greater than 0.999. Linearity was determined as follows: y = 0.0219x + 0.0025 and r 2 = 0.9999 for Phe, and y = 0.0168x + 0.0194 and r 2 = 0.9993 for Tyr (see Fig. S4 in the ESM). It should be noted that when ID-MS was applied, the y values represented the peak area ratio (analytes/ISTD). The endogenous concentration of Phe and Tyr in J0 were also determined by these calibration curves. Results showed a high sensitivity in comparison with the conventional PKU screening method and those of HPLC and MS/MS for amino acid profiling. The LOD and LOQ from the DBS were 0.03 and 0.10 mg/kg for Phe, and 0.07 and 0.25 mg/kg for Tyr, with signal-to-noise ratios of 3 and 10, respectively. This performance appears to be at least 10-fold more sensitive than previous results [3].
Recovery and reproducibility of DBS sampling were also evaluated from the experiment with the in-house calibrator. Recoveries were calculated by comparing the analytical values of J1–4 with the gravimetrically added amounts. The analytical values of J1–4 were obtained from the calibration curves above followed by subtraction of the J0 values, the endogenous levels. As indicated in Table 1, mean recoveries were close to 100 % where CVs were satisfactorily in the given concentration ranges. The recovery ranges of intra- and interday precisions from samples J1–4 were 98.3‒102.2 % (CV < 5.9 %) for Phe and 97.2‒101.6 % (CV < 6.2 %) for Tyr, respectively.
Clinical application and method comparisons
The proposed method was ultimately tested with clinical samples (107 PKU-negative and 14 PKU-positive blood spot specimens collected from Korean neonates), where two conventional ECA methods and the HPLC-MS method were applied simultaneously. As summarized in Table 2, diagnosis results based on two different diagnosis parameters agreed mostly with the decisions made from the BIA results. The diagnostic cutoff values for Phe concentration and Phe/Tyr ratio were 30 mg/L and 2.5, respectively [6]. The results of CE-MS and HPLC-MS were in excellent agreement, which supports the feasibility of the CE-MS method as a routine method for newborn screening. Among the few cases involving discrepancy of diagnosis results, we believe two false-positive cases were located by both the CE-MS and HLC-MS methods. Since the total number of positive cases was 14, the presence of two false-positive cases is not trivial (14 %). Since ECA and BIA are semiquantitative methods and prone to various interferences, it would be good practice to confirm the positive results with more robust methods such as CE-MS or HPLC-MS as demonstrated in this work. As is currently the case with HPLC-MS methods, CE-MS methods could develop and assume a greater role in quality control and quality assurance of conventional screening methods as they become more reliable for routine analysis.
Conclusions
In this study, we describe a simple, robust, highly selective, and high-sensitive CE-MS method for the direct analysis of Phe and Tyr in DBS. The sheathless CE-MS system was suitable for routine analysis in terms of operation stability, durability, and convenient system maintenance. Under optimized conditions, Phe and Tyr were determined within 3 min and exhibited good linearity and lower detection limits in DBS. We conducted experiments to overcome the analytical variation within DBS and between different DBS calibrators. Finally, we compared our method with conventional PKU screening methods, BIA, ECA, and HPLC-MS. The results showed good agreement with HPLC-MS and the potential of our method as a reference method for the use of ECA and BIA as routine analytical methods for clinical applications. While other methods (ECA, BIA, and HPLC, and MS) will continue to play a dominant role in this field, CE may evolve as an effective alternative due to its different separation mechanism, speed, high separation efficiency, and extremely low sample consumption. We hope our study provides the basis for such development.
References
Clague A, Thomas A (2002) Neonatal biochemical screening for disease. Clin Chim Acta 315(1–2):99–110
Senk P, Kozak L, Foret F (2004) Capillary electrophoresis and mass spectrometry for screening of metabolic disorders in newborns. Electrophoresis 25(10–11):1447–1456
Nagy K, Takats Z, Pollreisz F, Szabo T, Vekey K (2003) Direct tandem mass spectrometric analysis of amino acids in dried blood spots without chemical derivatization for neonatal screening. Rapid Commun Mass Spectrom 17(9):983–990
Kok WT, Brinkman UA, Frei RW (1983) Rapid determination of phenylalanine and tyrosine in urine and serum by HPLC with electrochemical detection. J Pharm Biomed Anal 1(3):369–372
Jeong JS, Sim HJ, Lee YM, Yoon HR, Lee DH, Hong SP (2009) Determination of phenylalanine in blood by high-performance anion-exchange chromatography-pulsed amperometric detection to diagnose phenylketonuria. J Chromatogr A 1216(30):5709–5714
Chace DH, Sherwin JE, Hillman SL, Lorey F, Cunningham GC (1998) Use of phenylalanine-to-tyrosine ratio determined by tandem mass spectrometry to improve newborn screening for phenylketonuria of early discharge specimens collected in the first 24 hours. Clin Chem 44(12):2405–2409
Tuchman M, McCann MT (1999) Phenylalanine and tyrosine quantification by stable isotope dilution liquid chromatography–mass spectrometry from filter paper blood spots. Clin Chem 45(4):571–573
Guthrie R, Susi A (1963) A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 32:338–343
Li W, Tse FL (2010) Dried blood spot sampling in combination with LC-MS/MS for quantitative analysis of small molecules. Biomed Chromatogr 24(1):49–65
Liu G, Ji QC, Jemal M, Tymiak AA, Arnold ME (2011) Approach to evaluating dried blood spot sample stability during drying process and discovery of a treated card to maintain analyte stability by rapid on-card pH modification. Anal Chem 83(23):9033–9038
Rago B, Liu J, Tan B, Holliman C (2011) Application of the dried spot sampling technique for rat cerebrospinal fluid sample collection and analysis. J Pharm Biomed Anal 55(5):1201–1207
Chace DH, Adam BW, Smith SJ, Alexander JR, Hillman SL, Hannon WH (1999) Validation of accuracy-based amino acid reference materials in dried-blood spots by tandem mass spectrometry for newborn screening assays. Clin Chem 45(8 Pt 1):1269–1277
Slazyk WE, Phillips DL, Therrell BL Jr, Hannon WH (1988) Effect of lot-to-lot variability in filter paper on the quantification of thyroxin, thyrotropin, and phenylalanine in dried-blood specimens. Clin Chem 34(1):53–58
Tamara T (1995) Inaccuracy in phenylalanine measurements—a problem towards acceptance of common screening approaches. Screening 4:5
Abdulrazzaq YM, Ibrahim A (2001) Determination of amino acids by ion-exchange chromatography on filter paper spotted blood samples stored at different temperatures and for different periods: comparison with capillary and venous blood. Clin Biochem 34(5):399–406
Adam BW, Alexander JR, Smith SJ, Chace DH, Loeber JG, Elvers LH, Hannon WH (2000) Recoveries of phenylalanine from two sets of dried-blood-spot reference materials: prediction from hematocrit, spot volume, and paper matrix. Clin Chem 46(1):126–128
Hanley WB, Demshar H, Preston MA, Borczyk A, Schoonheyt WE, Clarke JT, Feigenbaum A (1997) Newborn phenylketonuria (PKU) Guthrie (BIA) screening and early hospital discharge. Early Hum Dev 47(1):87–96
Schulze A, Mayatepek E, Hoffmann GF (2002) Evaluation of 6-year application of the enzymatic colorimetric phenylalanine assay in the setting of neonatal screening for phenylketonuria. Clin Chim Acta 317(1–2):27–37
Allard P, Cowell LD, Zytkovicz TH, Korson MS, Ampola MG (2004) Determination of phenylalanine and tyrosine in dried blood specimens by ion-exchange chromatography using the Hitachi L-8800 analyzer. Clin Biochem 37(10):857–862
Klampfl CW (2009) CE with MS detection: a rapidly developing hyphenated technique. Electrophoresis 30(Suppl 1):S83–91
Zamfir AD (2007) Recent advances in sheathless interfacing of capillary electrophoresis and electrospray ionization mass spectrometry. J Chromatogr A 1159(1–2):2–13
Shi LH, Jin YX, Moon DC, Kim SK, Park SR (2009) A sheathless CE/ESI-MS interface with an ionophore membrane-packed electro-conduction channel. Electrophoresis 30(10):1661–1669
Jeong JS, Kim SK, Park SR (2012) Capillary electrophoresis mass spectrometry with sheathless electrospray ionization for high sensitivity analysis of underivatized amino acids. Electrophoresis 33(14):2112–2121
Youhnovski N, Bergeron A, Furtado M, Garofolo F (2011) Pre-cut dried blood spot (PCDBS): an alternative to dried blood spot (DBS) technique to overcome hematocrit impact. Rapid Commun Mass Spectrom 25(19):2951–2958
Jeong JS, Lim HM, Kim SK, Ku HK, Oh KH, Park SR (2011) Quantification of human growth hormone by amino acid composition analysis using isotope dilution liquid-chromatography tandem mass spectrometry. J Chromatogr A 1218:6596–6602
Turgeon C, Magera MJ, Allard P, Tortorelli S, Gavrilov D, Oglesbee D, Raymond K, Rinaldo P, Matern D (2008) Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. Clin Chem 54(4):657–664
Jeong JS, Sim HJ, Lee YM, Yoon HR, Kwon HJ, Hong SP (2011) Chromatographic diagnosis of maple syrup urine disease by measuring the l-alloisoleucine/l-phenylalanine ratio in dried blood spots. J Chromatogr B Analyt Technol Biomed Life Sci 879(22):2171–2174
Dhondt JL, Loeber J, Elvers LH, Paux E (1998) Preparation of the first European working standard for phenylalanine determination in dried blood spots. J Med Screen 5(2):63–66
Dhondt JL, Paux E, Farriaux JP (1996) Need for a standardized procedure in the preparation of phenylalanine calibrators. Early Hum Dev 45(3):277–285
Acknowledgment
This work was supported by the Korea Research Institute of Standards and Science under the project “Development of Protein Measurement Standards,” Grant 13011017.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Amino Acid Analysis with guest editor Toshimasa Toyo'oka.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 401 kb)
Rights and permissions
About this article
Cite this article
Jeong, JS., Kim, SK. & Park, SR. Amino acid analysis of dried blood spots for diagnosis of phenylketonuria using capillary electrophoresis-mass spectrometry equipped with a sheathless electrospray ionization interface. Anal Bioanal Chem 405, 8063–8072 (2013). https://doi.org/10.1007/s00216-013-6999-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-6999-6