
Abstract
Edman degradation is a well-known method for obtaining amino acid (AA) sequences from a peptide by means of sequential reactions that release the N-terminal AAs from the peptide as a phenylthiohydantoin (PTH) derivative. Because of unexpected loss during the reaction and handling, there are few reports of use of this reaction for quantification. This manuscript describes the development of isotope-dilution liquid chromatography–electrospray ionization tandem mass spectrometry for 20 PTH-AA derivatives, and long-term stability testing of PTH-AAs to ensure quantitative quality in the reaction. The 20 corresponding [13C6]-PTH-AAs were prepared by use of a one-pot reaction involving a mixture of [13C6]-Edman reagent and 20 AAs. Good linearity was observed for standard curves for the PTH-AAs, using the corresponding [13C6]-PTH-AAs as internal standards (1–100 pmol per injection, r 2 = 0.989–1.000). Serum albumin (human), pepsin (porcine stomach mucosa), α-casein (bovine milk), ribonuclease A (bovine), lysozyme (chicken egg white), and insulin (bovine) subjected to Edman degradation were examined as model proteins and peptides for N-terminal AA analysis. The results of the impurity test were satisfactory. Yield from the entire reaction with human serum albumin was estimated to be at least 75 %, indicating great potential for absolute quantification of proteins without protein standards.

ᇵ
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Edman degradation, reported in 1950, is a well-known method for obtaining amino acid (AA) sequences from peptides [1]. The method is robust and reliable, and automated [2], miniaturized [3], strong chromogenic [4], and fluorescence [5, 6] methods have been reported to improve its application. At the Association of Biomolecular Resource Facilities (ABRF) the Edman Sequencing Research Group (ESRG), a currently active protein-sequencing research group (PSRG), has been working on practical problems using the method [7]; for example, the group applied the method to sequencing of proteins recovered from poly(vinylidene difluoride) (PVDF) membranes [8] and post-translationally modified peptides and proteins [9]. This traditional method has been widely used for protein identification via peptide sequences—even after mass spectrometry (MS)-based protein identification became popular, novel use of Edman degradation and its modified reagents for MS was reported to improve sequence analysis including ladder sequencing [10] by use of charged Edman type reagents [11–14] in combination with capping reagents [15].
The method consists of three reactions:
-
1.
coupling of Edman’s reagent (phenylisothiocyanate, PITC, Fig. 1a) with the N-terminal α-amino group to form a phenylthiocarbamoyl derivative (PTC-peptide, Fig. 1b);
Fig. 1 
Edman degradation. (a) Edman’s reagent (phenylisothiocyanate, PITC), (b) phenylthiocarbamoyl (PTC)-peptide, (c) anilinothiazolinone (ATZ)-AA, (d) des-1-peptide, (e) phenylthiohydantoin (PTH)-AA
-
2.
degradation of the PTC-peptide to produce the anilinothiazolinone derivative (ATZ-AA, Fig. 1c) and the des-1-peptide (Fig. 1d); and
-
3.
conversion of labile ATZ-AA into a more stable phenylthiohydantoin derivative (PTH-AA, Fig. 1e).

The resulting PTH-AA is then used to identify N-terminal AAs from their retention times by use of liquid chromatography–ultraviolet detection (LC–UV). Degradation can be reapplied to the remaining des-1 peptides (Fig. 1d) in the sample to identify the next AA, and the serial cycles enable sequencing from the N-terminus. It has been suggested that labeling of the N-terminal α-amino group is stoichiometric and that the subsequent degradation step to release the N-terminal is quantitative, but there are no reports available on quantitative aspects, especially for larger molecules, for example as intact proteins.
Quantification of proteins and peptides has recently been in high demand. In proteomics, quantification has been performed to estimate protein expression in some diseases. Of particular use is a combination of MS and isotope labeling, for example stable isotope labeling by AAs in cell culture [16], isotope-coded affinity tags (ICAT) [17], cleavable ICAT (cICAT) [18], and isobaric tags for relative and absolute quantification [19]. These labeling methods using isotope-coded reagents are becoming popular even for metabonomics [20], and have made major contributions to clinical chemistry. Furthermore, biopharmaceuticals have become popular in the pharmaceutical industry. Biotechnological and biological products, including protein and peptide-based drugs, require quality control on the basis of suggested guidelines, for example ICH-Q5E from the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Humans [21]. However, absolute quantification of an individual protein requires a sufficient amount of the corresponding pure standard. Therefore, absolute quantification has been achieved only when a limited number of proteins are targeted, for example amyloid betas in cerebrospinal fluid [22], membrane transporter proteins [23], and phosphorylation on the kinase activation loop of cellular focal adhesion kinase [24]. However, pure standard proteins are not always commercially available, and/or proteins are frequently supplied without information about their purity.
In 2007, Brune et al. reported quantitative data from Edman sequencing in an ABRF ESRG 2006 study from a peptide-quantification perspective [25]. They presented data for three peptides: peptide B, KAQYARSVLLEKDAEPDILELATGYR, peptide C, RQAKVLLYSGR, and peptide C*, RQAK(Ac)VLLYSGR. Absolute quantification from the first cycle gave 65.6 % and 67.0 % average recovery from peptides B and C + C*, respectively. Brune et al. could not explain this on the basis of their data, and said that because typical repetitive yields were 90 % to 98 %, the loss could not be attributed to overall coupling and cleavage reactions. This inspired the work discussed in this manuscript, in which an isotope-dilution method of Edman degradation was used to ensure quantitative quality in the reaction. Isotope dilution is a powerful method for performing accurate quantification, because the stable isotope analogues normalize for selective losses of analytes and act as carriers for trace amounts of analytes subjected to complex isolation procedures. When stable isotope analogues are used in combination with LC–selected reaction monitoring (SRM)-MS, it is possible to develop assays with specificity that cannot be achieved by use of any other technique [22, 26, 27].
Researchers in this laboratory had previously performed an experiment using matrix-assisted laser desorption-ionization (MALDI) time-of-flight (TOF)—MS to quantify bovine serum albumin as the PTH-aspartic acid (Asp) derived from the N-terminus, using [13C6]-PTH-Asp as the internal standard [28]. Therefore we decided to develop the isotope-dilution LC–electrospray ionization (ESI)—MS–MS method for 20 PTH-AA derivatives, for future quantitative application of the Edman degradation reaction. The corresponding 20 13C-labeled PTH-AAs were prepared as internal standards for the stable isotope-dilution technique. In addition to the work discussed here, several stable isotope-labeled Edman-type reagents have been reported [29–32]. However, all examples reported in proteomics are for use as tagging reagents for relative quantification of proteins and peptides [30–32]. Therefore, this work is the first isotope-dilution method for quantification of proteins by use of Edman’s reagent and including a degradation reaction. Because no information exists for PTH-AAs, long-term stability and/or storage conditions for the 20 PTH-AAs were also examined to compensate for unknown loss. Preliminary use to model proteins found that accurate N-terminal AA analysis was sufficient, at least for the impurity test, and the yield of the entire reaction with human serum albumin was estimated (by use of a manual liquid-phase procedure) to be at least 75 %. Therefore, this approach has great potential for absolute quantification, even for proteins without protein standards [33].
Materials and methods
Chemicals and materials
Ammonium formate, benzoic acid, ethyl acetate (AA sequence grade), ethyl mercaptan, formic acid (HPLC grade), 1-naphthaleneacetic acid, n-hexane, triethylamine, disodium hydrogen phosphate (dodecahydrate), sodium dihydrogen phosphate (dihydrate), trifluoroacetic acid (TFA), and hydrochloric acid (HCl, 0.1 mol L−1 aqueous solution) were purchased from Nacalai Tesque (Kyoto, Japan). N2 gas was purchased from Taiyo Nippon Sanso (Tokyo, Japan). Ar gas was purchased from Sendai Yozai (Sendai, Japan). PTH-serine (Ser), PTH-(N ε-PTC-)lysine (PTC-Lys), PTH-Asp, PTH-histidine (His) hydrochloride, PTH-S-carboxymethylcystein (cmCys), l-cmCys, l-asparagine (Asn) monohydrate, l-glutamine (Gln), l-tryptophan (Trp), phenol, phenyl isothiocyanate (PITC, AA sequence grade), AA mixed standard solution (AA sequence grade, type H without l-Asn, l-Gln and l-Trp; 2.5 mmol L−1 each in 0.1 mol L−1 aqueous HCl), and a mixture of PTH-AAs (AA sequence grade, without PTH-cmCys) were purchased from Wako Pure Chemical Industries (Osaka, Japan). PTH-alanine (Ala), PTH-arginine (Arg) hydrochloride, PTH-Asn, PTH-Gln, PTH-glutamic acid (Glu), PTH-glycine (Gly), PTH-isoleucine (Ile), PTH-leucine (Leu), PTH-methionine (Met), PTH-phenylalanine (Phe), PTH-proline (Pro), PTH-threonine (Thr), PTH-Trp, PTH-tyrosine (Tyr), and PTH-valine (Val) were purchased from Tokyo Chemical Industry (Tokyo, Japan). Acetonitrile (CH3CN, HPLC grade) was obtained from Kanto Chemical (Tokyo, Japan). Ethanol was purchased from Yamaichi Chemical Industries (Tokyo, Japan). Ultrapure water was obtained by use of a Milli-Q Integral 10 (EMD Millipore, Billerica, MA, USA) equipped with a 0.22 μm membrane cartridge. Serum albumin (human, 66 kDa), pepsin (porcine stomach mucosa, 35 kDa), α-casein (bovine milk, 23 kDa), ribonuclease A (bovine, 14 kDa), lysozyme (chicken egg white, 14 kDa), and insulin (bovine, 6 kDa) were purchased from Sigma–Aldrich (St Louis, MO, USA). [13C6]-PITC was prepared from [13C6]-aniline (Taiyo Nippon Sanso) with O,O′-di-2-pyridyl thiocarbonate (Tokyo Chemical Industry) in our laboratory, as described in our previous paper [27]. Microcentrifuge tubes (0.6 mL and 1.5 mL) were purchased from BM Equipment (Tokyo, Japan). Two-milliliter microcentrifuge tubes were purchased from As One (Osaka, Japan). A 5-μL microsyringe was purchased from Ito (Shizuoka, Japan). Microsyringes (25 μL and 500 μL) were purchased from Hamilton (Reno, NV, USA). Ammonium formate buffer (10 mmol L−1, pH 3), sodium phosphate buffer (5 mmol L−1, pH 7), and triethylamine-HCl buffer (5 mmol L−1, pH 11) were prepared in our laboratory.
LC–UV
LC was performed by use of a Shiseido Nanospace SI-1 2001 semimicro system (Shiseido, Tokyo, Japan). UV detection was performed by use of a Shiseido model 2002 UV–visible detector with Chromato-PRO ver. 2.0.2 (Runtime Instrument, Sagamihara, Japan) for data processing. An Inertsil ODS-4 column (150 mm × 2.1 mm i.d., 3 μm, 100 Å; GL Sciences, Tokyo, Japan) was used. Mobile phase component A was 0.1 % (v/v) aqueous formic acid–CH3CN (95:5, v/v), and mobile phase component B was 0.1 % (v/v) aqueous formic acid–CH3CN (5:95, v/v). For PTH-Asp, the linear gradient was: 5 % B at 0 min, 5 % B at 1 min, 35 % B at 15 min, 35 % B at 20 min, 95 % B at 25 min, 5 % B at 30 min, and 5 % B at 50 min, at a flow rate of 0.2 mL min−1 (program 1, Table S1, Electronic Supplementary Material). The linear gradient for the other PTH-Aas was: 5 % B at 0 min, 20 % B at 1 min, 35 % B at 15 min, 45 % B at 20 min, 45 % B at 35 min, 95 % B at 36 min, 95 % B at 40 min, 5 % B at 41 min, and 5 % B at 60 min, at a flow rate of 0.2 mL min−1 (program 2, Table S1, Electronic Supplementary Material).
LC–ESI–MS–MS
LC was performed by use of a Dionex Ultimate 3000 HPLC system (Dionex, Sunnyvale, CA, USA) with an Inertsil ODS-4 column (150 mm × 2.1 mm i.d., 3 μm, 100 Å; GL Sciences). Mobile phase component A was 0.1 % (v/v) aqueous formic acid–CH3CN (95:5, v/v), and mobile phase component B was 0.1 % (v/v) aqueous formic acid–CH3CN (5:95, v/v). The linear gradient was: 5 % B at 0 min, 15 % B at 2 min, 30 % B at 15 min, 50 % B at 40 min, 95 % B at 45 min, 95 % B at 55 min, 5 % B at 60 min, and 5 % B at 80 min, at a flow rate of 0.2 mL min−1 (program 3, Table S1, Electronic Supplementary Material).
ESI–MS–MS was performed by use of a TSQ Vantage (Thermo Fisher Scientific, Waltham, MA, USA), a triple-quadrupole mass spectrometer with ESI, operating in positive-ion mode. The optimum operating conditions were: source potential, 5 kV; heated capillary, 300 °C; sheath gas (N2), 60 arbitrary units; auxiliary gas (N2), 55 arbitrary units. For collision-induced dissociation (CID), Ar gas was used at a pressure of 1.0 mTorr, and the CID energy was optimized for each PTH-AA (Table 1). All MS data were processed by Qual Browser, a qualitative reviewing utility of Xcalibur ver. 2.1.0 (Thermo Fisher Scientific). Product-ion scanning of each PTH-AA was performed in infusion mode. Each PTH-AA solution (50 ng μL−1 in 1:1 (v/v) water–CH3CN) from a 500-μL microsyringe (10 μL min−1) was merged with the HPLC mobile phase (0.2 mL min−1, 30 % B) through a T-connector.
Stability of PTH-AAs
Four different solutions were tested for storage of the PTH-AAs: 25 % (v/v) aqueous TFA; 3:1 (v/v) 10 mmol L−1 ammonium formate buffer (pH 3)–CH3CN; 3:1 (v/v) 5 mmol L−1 sodium phosphate buffer (pH 7)–CH3CN; and 3:1 (v/v) 5 mmol L−1 triethylamine-HCl buffer (pH 11)–CH3CN. To avoid co-elution on the LC–UV chromatogram, mixtures of PTH-AAs were prepared as four different groups:
-
1.
PTH-Arg, PTH-Thr, PTH-Glu, PTH-Met, and PTH-Phe;
-
2.
PTH-Asn, PTH-Gly, PTH-cmCys, PTH-Pro, and PTH-PTC-Lys;
-
3.
PTH-His, PTH-Gln, PTH-Val, PTH-Trp, and PTH-Leu;
-
4.
PTH-Ser, PTH-Ala, PTH-Tyr, and PTH-Ile.
The sample of PTH-Asp, shown as a typical example, was prepared as a single sample. PTH-Asp, or each of the four mixtures of PTH-AAs (each 200 nmol), was transferred to a 2-mL microcentrifuge tube and evaporated under vacuum. Each residue was dissolved in 2.0 mL of the above buffers to give 100 μmol L−1 solution, and kept in the dark at 4 °C. For LC–UV analysis, 1-naphthaleneacetic acid (final concentration 100 μmol L−1, for groups 1–4, at pH 11), benzoic acid (final concentration 1 mmol L−1, for groups 1 and 2, in 25 % TFA), or phenol (final concentration 1 mmol L−1, for PTH-Asp in all the buffers, and groups 1 and 2 at pH 3 and pH 7, and groups 3 and 4 in 25 % TFA, pH 3 and pH 7) was used as internal standard. Aliquots (10 μL) of each solution were analyzed by use of LC–UV (269 nm, λ max [34]). Samples at pH 11 were neutralized by adding 0.1 mol L−1 aqueous HCl (4 μL) before injection. The [12C6]-PTH-AA-to-[13C6]-PTH-AA peak-area ratio was used for calculations. Peak areas at time zero were taken to be 100 % for normalization. Line graphs of recovery were plotted by use of Microsoft Excel 2007 work sheet ver. 12.0.
Preparation of 20 [13C6]-PTH-AAs
To prepare the stock samples, 4 μL AA mixture (2.5 mmol L−1 each in 0.1 mol L−1 aqueous HCl, no l-Asn, l-Gln, l-Trp) and 4 μL of a mixture of l-Asn, l-Gln, l-Trp, and l-cmCys (2.5 mmol L−1 each in 1:1 (v/v) water–CH3CN) were transferred to 0.6-mL microcentrifuge tubes and evaporated by use of a vacuum centrifuge (each sample 10 nmol per tube). Stock samples were kept in the dark at 4 °C until required. To prepare a mixture of [13C6]-PTH-AAs, 20 μL 100 mmol L−1 [13C6]-PITC in 8:1:1 (v/v) ethanol–water–triethylamine, freshly prepared) was added to each tube. After sealing N2 into the tube, the mixture was left to react at 55 °C on a heating block (HF-21; Yamato Scientific, Tokyo, Japan) for 30 min to form the [13C6]-PTC derivatives. After the reaction, the solution was evaporated by use of a vacuum centrifuge, and the residue was washed with 100 μL n-hexane and reconstituted with 50 μL 25 % (v/v) aqueous TFA. After sealing the N2 into the tube, the solution was left to react at 70 °C on a heating block for 30 min to form [13C6]-PTH-derivatives. The solution was evaporated by use of a vacuum centrifuge and kept as residue until required.
Calibration curves
Calibration curves were prepared by analysis of a mixture of AA solution and freshly prepared [13C6]-PTH-AA mixture as internal standard. Serial dilution AA mixtures (10, 20, 50, 100, 200, 500, or 1000 pmol each per 100 μL 3:1 (v/v) water–CH3CN) were prepared in a centrifuge tube from the above stock samples (10 nmol per tube). After evaporation by use of a vacuum centrifuge, the residue was left to react with PITC as described above. After the conversion reaction, 2 μL [13C6]-PTH-AA mixture (10 nmol each per 100 μL 3:1 (v/v) water–CH3CN, freshly prepared) was added to each tube as an internal standard mixture. The solution was evaporated by use of a vacuum centrifuge, and the residue redissolved in 100 μL 0.1 % (v/v) aqueous formic acid–CH3CN (3:1, v/v). An aliquot (10 μL) of each solution was subsequently analyzed by LC–ESI-SRM under the optimum SRM conditions shown in Table 1. To allow enough scanning time, multiple SRMs were divided into seven segments: t R 6–9.5 min (scan time 0.2 s), PTH-His; t R 9.5–12 min (scan time 0.1 s), PTH-Arg, Asn, Gln, Ser, and Thr; t R 12–14.5 min (scan time 0.1 s), PTH-Asp, Gln, Gly, Ser, and Thr; t R 14.5–19.5 min (scan time 0.16 s), PTH-Ala, Cys, and Glu; t R 19.5–22.5 min (scan time 0.2 s), PTH-Tyr; t R 22.5–27.5 min (scan time 0.16 s), PTH-Met, Pro, and Val; t R 27.5–35 min (scan time 0.125 s), PTH-Ile/Leu, Lys, Phe, and Trp. [12C6]-PTH-AA-to-[13C6]-PTH-AA peak-area ratio was used for calculation. Data analysis for calibration curves was performed by use of Quan Browser, a quantitative reviewing utility of Xcalibur ver. 2.1.0. The curves were plotted by use of simple linear regression without weighting and the origin was ignored.
Edman degradation of proteins
Each protein sample (500 pmol) was transferred to a microcentrifuge tube and evaporated under a N2 stream. Each residue was left to react with 50 μL PITC solution (7:1:1:1 (v/v) ethanol–water–triethylamine–PITC, freshly prepared) at 55 °C for 30 min under a N2 atmosphere. The reaction mixture was evaporated under a N2 stream, and the residue was then washed with 15:1 (v/v) n-hexane–ethyl acetate (3 × 100 μL). Ethyl mercaptan in TFA (0.1 %, v/v, 50 μL, freshly prepared) was added to the tube, which was then heated at 55 °C for 20 min. After evaporation under a N2 stream, ATZ-AAs in the residue were extracted by use of ethyl acetate (3 × 100 μL) and collected in a 0.6-mL microcentrifuge tube (total 300 μL). After evaporation under a N2 stream, the residue was redissolved in 50 μL 25 % (v/v) aqueous TFA and heated at 70 °C for 30 min. After the conversion reaction, 2 μL [13C6]-PTH-AA mixture (10 nmol each per 100 μL 3:1 (v/v) water–CH3CN, freshly prepared) was added to each tube as an internal standard mixture. The solution was evaporated by use of a vacuum centrifuge and the residue was redissolved in 100 μL 0.1 % (v/v) aqueous formic acid–CH3CN (3:1, v/v). An aliquot (10 μL) of each solution was then analyzed by use of LC–ESI-SRM as described above.
Results and discussion
Optimization of LC conditions
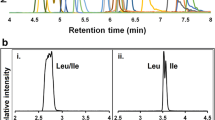
The conditions used for separation of the 20 PTH-AAs have already been optimized for LC–UV. Many of the conditions are not compatible with MS, however, for example when nonvolatile buffers, e.g. sodium acetate, are used [35] or when ion-pair reagents are added [36]. Because MS-based detection can discriminate overlapped peaks, we focused on separation of the two isomers PTH-Ile and PTH-Leu ([M + H]+ m/z 249) and their compatibility with ESI. The results showed that baseline separation between PTH-Ile and PTH-Leu could be achieved by use of 0.1 % (v/v) aqueous formic acid–CH3CN, which was also compatible with ESI (Fig. 2). Under these conditions, some PTH-AAs still overlapped (e.g. Arg with Asn, Thr with Gln, Ala with cmCys, Met with Val and Phe, Phe with Ile, and Leu with PTC-Lys); however, all overlapping peaks had different nominal masses. Conversely, PTH-His and [13C6]-PTH-Met share the same nominal mass ([M + H]+ m/z 273), but are well separated from each other on the chromatogram. Therefore, 20 PTH-AAs and 20 13C-labeled derivatives can be distinguished from each other under these conditions.

LC–ESI–MS chromatogram (total ion current) obtained from 20 PTH-AAs. Conditions are given in the “Materials and methods” section. PTH-AAs are abbreviated to three-letter AA names except Cys* (PTH-S-(carboxymethyl)-Cys) and Lys** (PTH-(N ε-phenylthiocarbamoyl(PTC))-Lys)
Optimization of SRM
MS–MS of individual PTH-AAs has been reported by Zhou et al. [37], but with different solvent conditions for the infusion study (1:1 (v/v) methanol–water containing 1 % acetic acid) from those in our study (0.1 % (v/v) aqueous formic acid–CH3CN). Therefore, we carefully examined product ion scanning of the 20 PTH-AAs for SRM, specifically in terms of reduced cross-talk between SRM channels. Typical fragmentation data from an acidic AA (PTH-Asp) and a basic AA (PTH-Arg) are shown in Fig. 3. Other examples are available in the electronic supplementary material (Fig. S1, Electronic Supplementary Material). MS–MS of PTH-Asp (m/z 251, [M + H]+) revealed one major product ion corresponding to [M + H − C4H5O3N1 (C6H5–NH=C=S)]+ at m/z 136, together with some small product ions, for example [M + H − C5H6O3N2S1 (C6H5)]+ at m/z 77, etc. MS–MS of PTH-Arg (m/z 292, [M + H]+) revealed four major product ions, m/z 233 ([M + H − guanidino group]+), m/z 174 (structure not identified), m/z 112 (structure not identified), and m/z 70 ([NH = CHCH2CH = CH2 + H]+). Furthermore, [C6H5–NH=C=S]+ at m/z 136, the most intense ion from PTH-Asp, was observed as a minor product ion. The results showed that protonation of PTH-derivatives from neutral and acidic AAs tended to occur on the thiohydantoin moiety, forming common product ions, for example m/z 136 (C6H5–NH+=C=S) and 77 (C6H5 +). In contrast, protonation of PTH derivatives from basic AAs tended to occur on the basic side chains, for example the imidazole ring of His or the guanidino group of Arg, forming characteristic product ions (i.e. m/z 152 from PTH-His, m/z 233 from PTH-Arg, etc). Product ions, including the phenyl group, were selected for SRM because of the labeling position of 13C in the ISs. Characteristic product ions were selected if possible; otherwise, the most intense product ions were selected (Table 1). Each optimum level of CID energy was determined from the breakdown curve in the auto-tune mode of the TSQ Vantage.

Typical MS–MS spectra obtained from PTH-AAs. (a) PTH-Asp (precursor ion, m/z 251), (b) PTH-Arg (precursor ion, m/z 292). Conditions: the CID energies for both PTH-Asp and PTH-Arg were 30 eV. Asterisks indicate product ions used for SRM
Preparation of 20 [12C6]-PTH-AAs and [13C6]-PTH-AAs
All PTH-AAs were originally commercially available. However, we decided to store the standards as more stable intact AA solutions because the long-term stability of PTH-AAs has not yet been reported. In addition, we believe that reacting AAs to form PTH-AAs with the samples for calibration curves can compensate for loss or deviation resulting from coupling and converting reactions and from extraction steps. Therefore, reaction conditions were optimized to prepare PTH-AAs (standards) and [13C6]-PTH-AAs (internal standards) from an AA mixture by reaction with PITC and [13C6]-PITC, respectively. The results showed that a two-step, one-pot reaction without a degradation step led to robust reproducibility for all PTH-derivatives. The SRM chromatogram from the reaction mixture of AA and PITC is shown in Fig. 4. The chromatogram from the reaction mixture of AA and [13C6]-PITC is available in the electronic supplementary material (Fig. S2, Electronic Supplementary Material). Cys was analyzed as the S-protected form (PTH-cmCys) in this reaction. Lys was analyzed as PTH-(N ε-PTC)-Lys because of the reactive ε-amino group on the side chain.

LC–ESI-SRM chromatogram obtained from 20 PTH-AAs. Conditions are given in the “Materials and methods” section. PTH-AAs are abbreviated to three-letter AA names except Cys* (PTH-S-(carboxymethyl)-Cys) and Lys** (PTH-(N ε-phenylthiocarbamoyl(PTC))-Lys)
Stability of PTH-AAs
Because the stability of PTH-AAs has not yet been reported, we decided to store the standards as intact AA solutions. However, to further improve quantification, stability was examined in acidic, neutral, and basic conditions—10 mmol L−1 ammonium formate buffer (pH 3)–CH3CN (3:1, v/v); 5 mmol L−1 sodium phosphate buffer (pH 7)–CH3CN (3:1, v/v); and 5 mmol L−1 triethylamine-HCl buffer (pH 11)–CH3CN (3:1, v/v), respectively. CH3CN was added to all buffers to improve solubility. The reaction solution for PTH-AA conversion (25 % (v/v) aqueous TFA) was also examined. PTH-AAs were dissolved in these buffers and the resulting solutions (100 μmol L−1) were kept at 4 °C.
Peaks from decomposed products can interfere on LC–UV. Therefore, we prepared test mixtures of PTH-AAs as four different groups to avoid co-elution. 1-Naphthalenesulfonic acid, benzoic acid, or phenol was added to the solutions as the internal standard. Typical examples from the acidic (PTH-Asp) and basic (PTH-Arg) AAs are shown in Fig. 5. Other examples are available in the electronic supplementary material (Fig. S3, Electronic Supplementary Material). All PTH-AAs decomposed quickly under basic conditions (pH 11). PTH-Arg, cmCys, Gln, His, Leu, Ser, and Thr were highly unstable (less than 50 % remaining after four weeks) under neutral conditions (pH 7). Acidic conditions (pH 3 and 25 % aqueous TFA) were most favorable for almost all PTH-AAs, but substantial amounts of PTH-Ser and Gln were lost (less than 50 % remaining after four weeks) in this buffer (pH 3). As a result, 25 % (v/v) TFA was thought to be the most suitable solution for storing PTH-AAs. Because the last conversion reaction of ATZ-AAs to PTH-AAs was performed in this solution, it was logical to leave the PTH-AAs in the reaction mixture. Because PTH-AA standards were freshly prepared each time, with the test samples, loss as a result of instability was negligible.

Typical stability of PTH-AAs: (a) PTH-Asp, (b) PTH-Arg. Conditions: (circles, n = 3) 25 % (v/v) aqueous TFA, (squares, n = 3) 10 mmol L−1 ammonium formate buffer (pH 3)–CH3CN (3:1, v/v), (triangles, n = 3) 5 mmol L−1 sodium phosphate buffer (pH 7)–CH3CN (3:1, v/v), (diamonds, n = 1) 5 mmol L−1 triethylamine-HCl buffer (pH 11)–CH3CN (3:1, v/v). PTH-AAs were dissolved in these buffers at 100 μmol L−1 and stored at 4 °C
Calibration curve obtained by plotting [12C6]-PTH-AA-to-[13C6]-PTH-AA peak-area ratio
Calibration curves were prepared from AA mixtures with the corresponding [13C6]-PTH-AAs as internal standards (Table S2, Electronic Supplementary Material). Good linearity was observed for all the calibration curves over the entire range (1–100 pmol per injection, r 2 = 0.989–1.000). Because the calibration curves were prepared from an AA mixture (Fig. 6b), together with specimens (Fig. 6a) and added [13C6]-PTH-AAs (internal standards) (Fig. 6c), compensation for errors arising from the coupling and stabilization reactions and for errors arising from instability and/or extraction efficiency of PTH-AAs was achieved.

The entire procedure for isotope-dilution LC–ESI–MS–MS for the PTH-AA derived from peptides or proteins. Samples for the calibration curve (b) were prepared with the specimens (a) to compensate for errors from coupling and stabilization reactions. [13C6]-PTH-AAs (internal standards) were added to samples for the calibration curve (b) and to the specimen (a) to compensate for the errors from future degradation
Applications to proteins
Edman degradation was originally used for peptides, not large proteins. Therefore, this approach was first applied to six model proteins—serum albumin (human, Fig. S4a, Electronic Supplementary Material), α-casein (bovine milk, Fig. S4b, Electronic Supplementary Material), lysozyme (chicken egg white, Fig. S4c, Electronic Supplementary Material), ribonuclease A (bovine, Fig. S4d, Electronic Supplementary Material), pepsin (porcine stomach mucosa, Fig. S4e, Electronic Supplementary Material), and insulin (bovine, Fig. S4f, Electronic Supplementary Material)—to verify their suitability. Corresponding N-terminal AAs were released from all model proteins as PTH-AAs. Because unexpected PTH-AAs were found as precursor proteins and/or self-digested proteins (from ribonuclease A, Fig. S4d, and pepsin, Fig. S4e; Electronic Supplementary Material), this method might be useful for identifying impurities in standard proteins. Furthermore, PTH-Gly and PTH-Phe were found in insulin (Fig. S4f, Electronic Supplementary Material) as a result of the A-chain and B-chain, respectively. Because applicability to large molecules depends on the yield of the entire process, we performed serial Edman degradation using human serum albumin, which was the largest model protein (66 kDa) examined (Table 2). From the first cycle, the amount of N-terminal Asp1 found was 18.3 pmol, together with Ala2 (0.84 pmol) and His3 (2.39 pmol), which are believed to be from N-truncated human serum albumin. To confirm recovery, further cycles were performed on the same samples. The second and third cycles gave 2.81 pmol and 2.53 pmol, respectively. The reason for incomplete recovery is still under investigation, but we postulate that the efficiency of extraction of ATZ-AAs from coagulated protein must be improved. However, we achieved 77 % (18.3/18.3 + 2.81 + 2.53) recovery from the large protein despite use of a manual liquid-phase procedure; therefore, this method could have great potential for quantification of proteins without standards.
Conclusion and outlook
Although Edman sequencing has been widely used, there is a lack of studies relating to quantification. As Brune et al. suggest [24], we believe that the problems of quantification should be attributed not to overall coupling and cleavage reactions but to something else, for example extraction yields and/or the instability of PTH-AAs. This research found that stable isotope-dilution LC–ESI–MS–MS compensated as much as possible for loss in the determination of 20 PTH-AAs. This research also demonstrated the previously unreported long-term stability of 20 PTH-AAs. Because this strategy can improve the quality of quantification, the objective of this research was to apply this method as a breakthrough in the search for absolute quantitative proteomics [33]. We believe that PTH-AAs derived from Edman degradation of proteins have great potential for use in absolute quantification of proteins without protein standards.
References
Edman P (1950) Method for determination of the amino acid sequence in peptides. Acta Chem Scand 4:283–293
Edman P, Begg G (1967) A protein sequenator. Eur J Biochem 1:80–91
Chen W, Yin X, Mu J, Yin Y (2007) Subfemtomole level protein sequencing by Edman degradation carried out in a microfluidic chip. Chem Commun (Camb) 2488–2490
Chang JY, Creaser EH, Bentley KW (1976) 4-NN-dimethylaminoazobenzene 4′-isothiocyanate, a new chromophoric reagent for protein sequence analysis. Biochem J 153:607–611
Jin SW, Chen GX, Palacz Z, Wittmann-Liebold B (1986) A new sensitive Edman-type reagent: 4-(N-1-dimethylaminonaphthalene-5-sulfonylamino)phenyl isothiocyanate. Its synthesis and application for micro-sequencing of polypeptides. FEBS Lett 198:150–154
Muramoto K, Nokihara K, Ueda A, Kamiya H (1994) Gas-phase microsequencing of peptides and proteins with a fluorescent Edman-type reagent, fluorescein isothiocyanate. Biosci Biotechnol Biochem 58:300–304
http://www.abrf.org/index.cfm/group.show/EdmanSequencing.29.htm
Buckel S, Cook R, Crawford J, Dupont D, Madden B, Neveu J, Steinke L, Fernandez J (2002) ABRF-2002ESRG, a difficult sequence: analysis of a PVDF-bound known protein with a heterogeneous amino-terminus. J Biomol Tech 13:246–257
Mische SM, Yüksel KÜ, Mende-Mueller LM, Matsudaira P, Crimmins DL, Andrews P (1993) Protein sequencing of post-translationally modified peptides and proteins: Design, characterization and results of ABRF-92SEQ. In: Hogue-Angeletti R (ed) Techniques in protein chemistry IV. Academic Press Inc., San Diego, pp 453–461
Chait BT, Wang R, Beavis RC, Kent SBH (1993) Protein ladder sequencing. Science 262:89–92
Aebersold R, Bures EJ, Namchuk M, Goghari MH, Shushan B, Covey TC (1992) Design, synthesis, and characterization of a protein sequencing reagent yielding amino acid derivatives with enhanced detectability by mass spectrometry. Protein Sci 1:494–503
Bures EJ, Nika H, Chow DT, Morrison HD, Hess D, Aebersold R (1995) Synthesis of the protein-sequencing reagent 4-(3-pyridinylmethylaminocarboxypropyl) phenyl isothiocyanate and characterization of 4-(3-pyridinylmethylaminocarboxypropyl) phenylthiohydantoins. Anal Biochem 224:364–372
Ducret A, Bures EJ, Aebersold R (1997) High-sensitivity detection of 4-(3-pyridinylmethylaminocarboxypropyl) phenylthiohydantoins by capillary liquid chromatography–microelectrospray ion trap mass spectrometry. J Protein Chem 16:323–328
Marekov LN, Steinert PM (2003) Charge derivatization by 4-sulfophenyl isothiocyanate enhances peptide sequencing by post-source decay matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J Mass Spectrom 38:373–377
Thakkar A, Wavreille AS, Pei D (2006) Traceless capping agent for peptide sequencing by partial Edman degradation and mass spectrometry. Anal Chem 78:5935–5939
Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1:376–386
Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 17:994–999
Li J, Steen H, Gygi SP (2003) Protein profiling with Cleavable Isotope-Coded Affinity Tag (cICAT) reagents. Mol Cell Proteomics 2:1198–1204
Zieske LR (2006) A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J Exp Bot 57:1501–1508
Leng J, Wang H, Zhang L, Zhang J, Wang H, Guo Y (2013) A highly sensitive isotope-coded derivatization method and its application for the mass spectrometric analysis of analytes containing the carboxyl group. Anal Chim Acta 758:114–121
Guidance for Industry, Q5E Comparability of biotechnological/biological products subject to changes in their manufacturing process, June 2005, ICH. (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073476.pdf)
Oe T, Ackermann BL, Inoue K, Berna MJ, Garner CO, Gelfanova V, Dean RA, Siemers ER, Holtzman DM, Farlow MR, Blair IA (2006) Quantitative analysis of amyloid beta peptides in cerebrospinal fluid of Alzheimer’s disease patients by immunoaffinity purification and stable isotope dilution liquid chromatography/negative electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom 20:3723–3735
Kamiie J, Ohtsuki S, Iwase R, Ohmine K, Katsukura Y, Yanai K, Sekine Y, Uchida Y, Ito S, Terasaki T (2008) Quantitative atlas of membrane transporter proteins: development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm Res 25:1469–1483
Ciccimaro E, Hanks SK, Yu KH, Blair IA (2009) Absolute quantification of phosphorylation on the kinase activation loop of cellular focal adhesion kinase by stable isotope dilution liquid chromatography/mass spectrometry. Anal Chem 81:3304–3313
Brune DC, Hampton B, Kobayashi R, Leone JW, Linse KD, Pohl J, Thoma RS, Denslow ND (2007) ABRF ESRG 2006 study: Edman sequencing as a method for polypeptide quantitation. J Biomol Tech 18:306–320
Oe T, Kambouris SJ, Walker VE, Meng Q, Recio L, Wherli S, Chaudhary AK, Blair IA (1999) Persistence of N7-(2,3,4-trihydroxybutyl)guanine adducts in the livers of mice and rats exposed to 1,3-butadiene. Chem Res Toxicol 12:247–257
Oe T, Tian Y, O’Dwyer PJ, Roberts DW, Malone MD, Bailey CJ, Blair IA (2002) A validated liquid chromatography/tandem mass spectrometry assay for cis-amminedichloro(2-methylpyridine)platinum(II) in human plasma ultrafiltrate. Anal Chem 74:591–599
Oe T, Maekawa M, Satoh R, Lee SH, Goto T (2010) Combining [13C6]-phenylisothiocyanate and the Edman degradation reaction: a possible breakthrough for absolute quantitative proteomics together with protein identification. Rapid Commun Mass Spectrom 24:173–179
Ares JJ, Fowble JW, Urchek TG, Miller DD (1987) Synthesis and application of 13C enriched phenylisothiocyanate as a 13C NMR protein probe. Biochem Biophys Res Commun 142:1064–1071
Guillaume E, Panchaud A, Affolter M, Desvergnes V, Kussmann M (2006) Differentially isotope-coded N-terminal protein sulphonation: combining protein identification and quantification. Proteomics 6:2338–2349
Leng JP, Wang HY, Zhang L, Zhang J, Wang H, Cai TT, Yao JT, Guo YL (2011) Integration of high accuracy N-terminus identification in peptide sequencing and comparative protein analysis via isothiocyanate-based isotope labeling reagent with ESI ion-trap TOF MS. J Am Soc Mass Spectrom 22:1204–1213
Leng J, Zhu D, Wu D, Zhu T, Zhao N, Guo Y (2012) Analysis of the differentially expressed low molecular weight peptides in human serum via an N-terminal isotope labeling technique combining nano-liquid chromatography/matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom 26:2555–2562
Anonymous (2010) Novel approach to absolute protein quantitation. Expert Rev Proteomics 7:13–13
Smith GF, Murray M (1968) Direct spectrophotometric quantitation of phenylthiohydantoin derivatives of amino acids from thin layers of silica gel. Anal Biochem 23:183–195
Glajch JL, Gluckman JC, Charikofsky JG, Minor JM, Kirkland JJ (1985) Simultaneous selectivity optimization of mobile and stationary phases in reversed-phased liquid chromatography for isocratic separations of phenylthiohydantoin amino acid derivatives. J Chromatogr A 318:23–39
Tsunasawa S, Kondo J, Sakiyama F (1985) Isocratic separation of PTH-amino acids at picomole level by reverse-phase HPLC in the presence of sodium dodecylsulfate. J Biochem 97:701–704
Zhou J, Hefta S, Lee TD (1997) High sensitivity analysis of phenylthiohydantoin amino acid derivatives by electrospray mass spectrometry. J Am Soc Mass Spectrom 8:1165–1174
Acknowledgments
This work was supported in part by the Asahi Glass Foundation through a grant to T.O. (2010–2011, “Novel analytical platform to enable both absolute quantification and identification of unknown proteins in biological samples”) and through their continuing support for the restoration of our laboratories damaged by the Great East Japan Earthquake. The authors also thank Shionogi & Co., Ltd. (Osaka, Japan), Astellas Pharma Inc. (Tokyo, Japan), and Professor Ian A. Blair (University of Pennsylvania, Philadelphia, PA) for donating two used TSQ 7000 s and an LCQ Deca, respectively.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Amino Acid Analysis with guest editor Toshimasa Toyo’oka.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1.63 MB)
Rights and permissions
About this article
Cite this article
Satoh, R., Goto, T., Lee, S.H. et al. Can Edman degradation be used for quantification? Isotope-dilution liquid chromatography–electrospray ionization tandem mass spectrometry and the long-term stability of 20 phenylthiohydantoin-amino acids. Anal Bioanal Chem 405, 8001–8010 (2013). https://doi.org/10.1007/s00216-013-6901-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-013-6901-6