
Abstract
By the example of electrodeposition of Co–W alloys, this work shows that observed peculiarities of induced codeposition, including the macroscopic size effect in the composition and properties of deposited layers and their nanocrystallinity, are a consequence of the fact that the deposition-inducing species (a complex of the deposition-inducing metal) has the form of a high-molecular-weight polymer. Under the conditions of (relatively) high current loading on a plating electrolyte (high volume current density), this results in involvement of water molecules in the electrochemical process, formation of oxy-hydroxide layers, hydrogenation, an increase in the alloy tungsten content as a result of the side reaction of hydrogen evolution, alkalization of near-electrode region, and polymerization of the deposition-inducing metal species. Because of the presence of macroscopic size effect (i.e., the dependences of composition and properties of deposited coatings on the electrodeposition surface area), industrial scaling up of this electrodeposition technology will require maintaining the current loading on a plating bath at a constant level, along with other parameters traditional for electrochemical materials science.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The interest in developing methods for electrochemical deposition of alloys of iron group metals with refractory metals (W, Mo, Re) is stimulated by their good microhardness, corrosion resistance, catalytic performance toward the hydrogen evolution reaction (HER), and the possibility to control their magnetic behavior [1–3]. Methods for preparation of thin films and quasi-one-dimensional structures (nanotubes, nanowires, nanorods) of these alloys were reported [4, 5]. The electrodeposition mechanism of such coatings, however, is still a subject of debates [1, 2, 6, 7]. Electrodeposition of these alloys was categorized as anomalous [1], because the traditional electrochemical methods for control of alloy composition (and properties) are not applicable here. Further, this kind of electrodeposition was defined as induced codeposition [8], where a complex of an iron group metal induces deposition of a refractory metal to form an alloy. All mechanisms of these processes known to date can be divided into two groups depending on whether deposition involves a mixed complex (pre-)formed in solution [1, 9] or it is mediated by the interaction on the surface between intermediates resulting from partial reduction of iron group metal complexes and a refractory metal ([2] and references therein, as well as [7, 10–12]).
In many studies, these processes are considered to be catalytic [11–14], with the catalysis being carried out by a reduced form of the deposition-inducing metal species [13], or a complex of the deposition-inducing metal [11, 12], or the deposited alloy (which is catalytically active toward HER [14]). In some studies, the side process of hydrogen evolution is thought to be a dominant process [2]. Since electrodeposition of the considered alloys normally involves HER, the current efficiency of the target process may be well below 100%, and experiments demonstrated that the formation of this kind of alloys with a high refractory metal content correlates with a low current efficiency [15].
Macroscopic size effects (MSEs) in properties and composition of coatings prepared by induced codeposition, which are undoubtedly related to the deposition mechanism, must be considered as important features of this type of coatings [16–23]. These effects imply that the composition and properties of prepared coatings change when varying the electrodeposition surface area while keeping the electrolyte composition, volume, and pH, as well as the deposition temperature and current density unchanged. We note that the considered effect was observed not only when the electrodeposition current density was held constant [16, 18–22] but also in potentiostatic experiments [17, 23]. It was demonstrated that there may be different pathways to the formation of surface layers [23].
It was illustrated [16–21] (and presented in a generalized form [22]) that the formation of surface oxy-hydroxide layers (the extent of hydrogenation, thickness, structure, and composition depending on volume current density (VCD)) result in a decrease in surface microhardness (Fig. 1). We note that VCD can be considered as a measure of current loading on an electrolyte. For a definite electrodeposition current density, an increase in VCD contributes to a decrease in microhardness. In conflict with this, the presence of an oxide on the surface contributes to its enhanced corrosion resistance. Consequently, raising the VCD, i.e., the current loading on the electrolyte, at a definite electrodeposition current density and bath volume is equivalent to increasing the electrodeposition surface area, which leads to lower corrosion current density of the resulting surface [21].

Effect of volume current density (VCD) on the microhardness of the surface of deposited coating [17].
In addition, an increase in the current loading on the electrolyte at a definite current density was shown to result in the higher potential, lower current efficiency, and an increase in the alloy tungsten content (Fig. 2). Put differently, the electrode potential, current efficiency, and the coating composition depend on the electrodeposition surface area at a given value of electrodeposition current density [23]. This can create considerable technological challenges in industrial scaling up of the considered process and its application to coating of both large and small (micromachining) surfaces. And the VCD can be considered as a measure of current loading on the electrolyte.

Effect of VCD on (1) tungsten content of formed Co–W alloys, (2) potential, and (3) current efficiency (CE) observed for electrodeposition from the citrate bath at a current density of 2 A/dm2 [23].
Thus, so far we have identified two variants of MSEs affecting the coatings prepared by induced codeposition: (1) the effect of electrodeposition surface area and VCD on the composition and properties of the surface layer; and (2) alteration of the ratio between alloying components (i.e., the deposition-inducing metal and the refractory metal) in the bulk of a deposited coating. At the same time, an increase in the VCD may have different effects on microhardness. While the increase in VCD in the first variant results in a decrease in microhardness, in the second variant, the content of refractory metal in the alloy grows, which must contribute to an increase in microhardness.
The observed effects, however, cannot be predicted from the mechanisms of induced codeposition known to date [1, 2, 6, 7, 11–14]. It can be expected that this gap stems from that fact that these mechanisms are electrochemical and they do not allow for (to the required extent) the composition and structure of complexes present in plating baths. In addition to that, our earlier studies [9, 24, 25] demonstrated that the considered electrolytes contain high-molecular-weight complexes, their molecular weight exceeding that of B12 vitamin; and study [21] found that the structures of such agglomerated complexes can be conveniently represented by mathematical models for fractal systems.
The present work is concentrated on the experimental investigation of the effect that the current loading on electrolyte has on properties of metal coatings obtained by induced codeposition (by the example of deposition of Co–W alloys from the citrate bath) and aims at determining pathways leading to their formation, which will facilitate development of methods for controlling their composition and properties as a result. We highlight that this kind of effect was observed only for induced codeposition of iron group metals with tungsten [16–23].
EXPERIMENTAL
Presented below are results of the study of composition of Co–W coatings deposited from a citrate electrolyte under various conditions, as well as the results of electrolyte analysis performed during electrodeposition. The bath composition and temperature are provided in Table 1.
The electrodeposition was conducted under galvanostatic conditions at current densities varying in the range of 1–5 A/dm2 and under potentiostatic conditions at potentials of –0.7 and –1.1 V vs a Ag/AgCl (sat) reference electrode. The process was performed using an Autolab PGSTAT302N potentiostat/galvanostat (Metrohm). The coatings were deposited from both quiescent solutions and solutions stirred with a magnetic stirrer. A series of experiments was performed on a 3-mm platinum disk electrode using rotating disk electrode (RDE) technique. The electrode surface was subjected to the following anodic–cathodic treatment in 35% nitric acid before measurements: an anodic potential of +1 V was applied until the current fell below 0.0002 A, followed by a cathodic potential of –0.2 V, until the cathodic current reached a peak and started to decrease. Since the composition and properties depend on VCD (I/V), as was mentioned above, this parameter was varied from 20 to 400 mA/L by varying both the electrodeposition surface area from 0.5 to 8 cm2 and the electrolyte volume from 50 mL to 1 L. The electrodepositions were performed on copper substrates on which a Ni strike layer (thickness, ~0.5 µm) was electroplated from a nickel plating bath by applying a current density of 30 mA/cm2 for 60 s. The electrodepositions were carried out in a single-compartment cell using non-consumable (graphite) anodes.
The conditions for electrodeposition and dissolution of deposited coatings were established in preliminary cyclic voltammetry (CV) experiments in a three-electrode cell, using a Pt working electrode, a 1 cm2 Pt flag counter electrode, and a Ag/AgCl (sat) reference electrode. The experiments were conducted at 80°C and a potential scan rate of 10 mV/s. RDE polarization curves were recorded at different rotation rates, and electrochemical impedance spectroscopy was used to investigate the structure of resulting surface layers.
The coating composition was determined by X-ray fluorescence (XRF) analysis on an X-Calibur spectrometer (Xenemetrix). The variant of XRF used in this study allowed us to determine only the heavy elements Co, W, Cu, and Ni, the latter two being the substrate elements. Normally, because the prepared coatings were sufficiently thick (20–70 μm), only Co and W were detected and their alloy content (wt %) was quantified.
Because our earlier studies identified the rate of change in the bulk concentration of the deposition-inducing metal complex as one of the causes of the MSE [19–21], electronic absorption spectra of the considered citrate bath were recorded during electrodeposition using a Specord M40 UV–Vis spectrometer.
The current efficiency was determined by measuring the coating mass while also taking into account the alloy content of the heavy elements. The durations of both galvanostatic and potentiostatic depositions were 0.5–1.5 h.
RESULTS AND DISCUSSION
Electrode Processes and the Surface Structure
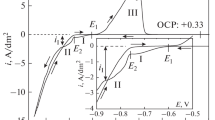
Two signature regions denoted by I and II can be identified in the cathodic branch of a polarization curve recorded in the considered electrolyte (Fig. 3). Radically different electrode processes occur in these regions depending on hydrodynamic conditions. While the current density in region II grows with increasing the rotation rate, an anomalous decrease in the current density is observed in region I, as the frequency of RDE rotation increases. It is to be noted that a similar decrease was observed in reduction of an iron–citrate complex present in the electrolyte lacking sodium tungstate [26]. Causes of such an effect must be addressed in a more detailed investigation.

Potentiodynamic curves in the citrate bath for induced codeposition of Co–W alloy: (1) RDE, 200 rpm; (2) RDE, 2000 rpm; and (3) Pt wire electrode, under quiescent conditions. Temperature, 80°C; potential scan rate, 10 mV/s.
Admittedly, potential region I is the region preceding the alloy deposition, because the alloy is not formed at these potentials (the peak of anodic dissolution is lacking when scanning to –0.76 V). As can be seen in Figs. 3 and 4, the alloy is formed at a cathodic potential negative of –0.76 V (denoted by E2 in Fig. 4). The rise in current density is observed specifically on reaching this potential, which corresponds to cobalt(II) citrate reduction, and the reverse anodic branch of the CV curve features the signature peak of anodic dissolution at potential E3 (Fig. 4), which is not observed at potentials corresponding to I.

CV curves on a Pt wire electrode in the citrate bath at 80°C under quiescent conditions; scan rate, 10 mV/s.
Region II is the region of mixed kinetics where the kinetic current constitutes a considerable fraction of the overall current (Fig. 5). This suggests that a good analysis of voltammetry data requires taking into account not only the geometric but also the true surface area when calculating the current density.

Koutecky–Levich plot for a potential of –1.0 V.
As was seen in study [26] by the example of deposition of Fe–W alloys, region I represents the reduction of tungstate to an intermediate oxidation state, the degree of oxidation depending on the potential. The oxy-hydroxide tungsten-containing layers which determine the observed results were obtained by carrying out electrodeposition at a potential of –0.7 V for 10 min, and EIS spectra were then recorded at the OCP. EIS plots obtained at different RDE rotation rates are presented in Fig. 6. The equivalent circuit representing the EIS data of the present study (Fig. 6b) was borrowed from our earlier work on electrodeposition of the Co–W alloy from the citrate bath [27]. The circuit includes the double-layer capacitance CPE1; the combined resistance of the porous and the oxy-hydroxide layers and the electrolyte in its pores, R2; the capacitance CPE2 associated with a process at the oxy-hydroxide layer (most probably to be adsorption); and charge transfer resistance R3 related to this process (Fig. 6b). Significant are the facts that (i) the capacitance CPE1 does not change (within ±2.5%) with the rotation rate in the range of 0–2000 rpm, which is intuitive for common double-layer dimensions; and (ii) the value for CPE1 (87 μF/cm2) is at least 1.5 times higher normal values for electrical double layer capacitance (20–60 μF/cm2). As for the charge separation region CPE2, its capacitance is several times that of CPE1 and it displays an increasing dependence on the rotation rate (Table 2). The implication of this fact is that this region is so large that it is influenced by tangential stresses within the hydrodynamic layer. In addition, resistance R3 grows with increasing the rotation rate, which results in a decrease in the current density with increasing RDE rotation rate (Fig. 3). Since the equivalent circuit in Fig. 6b implies the presence of an adsorbed layer on the electrode surface [27], it should be admitted that the absorbed complex has a high molecular weight.

(a) EIS graphs recorded on the RDE at different rotation rates at the OCP in the considered electrolyte after poising the electrode at –0.7 V for 10 min: (1) 0, (2) 200, and (3) 2000 rpm. (b) Proposed equivalent circuit.
Earlier, it was noted that the effect that VCD, i.e., the current loading on the electrolyte, has on the coating composition and properties is a consequence of the high rate of change in the concentration of deposition-inducing metal complex, which is an iron group metal complex [22]. And on the other hand, this occurs when the analytical concentration of the iron group metal is considerably higher than the concentration of iron group metal complex. That is, we are bound to assume that the deposition-inducing metal complex is a three-dimensional polymer-like structure with a high molecular weight. Earlier studies demonstrated that iron–citrate complexes in neutral or alkaline media (pH > 4) form polymers with a molecular weight of ~ 2 × 105 [28, 29]. It was also noted that these polymers have a linear chain structure [28, 29]. For a cobalt sulfate molar concentration of 0.2 M (Table 1), the concentration of the complex is ~ 1.5 × 10–4 M. At this concentration of deposition-inducing complex, the current loading must have an effect on the electrolyte, with the extent of this effect increasing with increasing the electrodeposition surface area, because the current loading grows as the electrodeposition surface area is increased.
It seems reasonable to assume that this also applies to Co citrate complexes. It should be taken into account that the sequence in which the electrolyte components are combined is an important factor in the formation of polymers with a structure specified in [28, 29]. Thus, sodium tungstate is added (as a solid or in a dissolved form) last, and considerable local alkalization occurs during mixing or dissolution which results in observable precipitation of cobalt hydroxide, which subsequently dissolves to form citrate complexes or colloidal particles. Alkalization of the near-electrode region during induced codeposition of the alloy has similar effect. The value of different fractions of the citrate electrolyte (those with higher and lower molecular weights) in deposition of Co–W alloys [24, 25] will be addressed later in the text.
As a result, the anomalous effect of hydrodynamics on the capacitance CPE2 (charge separation region) at potentials preceding the alloy deposition region, as well as the diminution of current density when increasing the rotation rate, are consequences of adsorption of the high-molecular-weight complexes (Co-citrate and W-citrate) on the electrode surface.
MACROSCOPIC SIZE EFFECTS AS A CONSEQUENCE OF THE RATE OF CHANGE IN THE BULK CONCENTRATION OF DEPOSITION-INDUCING METAL COMPLEX
MSEs affecting the coating elemental composition and properties (i.e., the influence of electrodeposition surface area on the coating composition and properties) were observed for coatings prepared by induced codeposition under galvanostatic conditions [16–23], but they were also detected in potentiostatic depositions (Fig. 7). The cause of the observed effect (i.e., a decrease in current density when increasing the electrodeposition surface area) is related to the mixed kinetics of electrodeposition process, according to which the current density per geometric surface area must decrease as the surface area increases, because the density of kinetic current is determined by the true, not geometric, surface area. For instance, for a roughness factor of ~10, a 5-fold increase in the apparent surface area corresponds to a 50-fold increase in the true area. Since the measured current is due to the mixed kinetic-diffusion control, the current density calculated per apparent surface area decreases during prolong electrolysis (~1 h) by only a factor of two (see the results for surface areas of 1 and 5 cm2 in Fig. 7). It is to be noted that such a decrease was observed only in relatively long electrolysis, when the coating thickness reached ~15 µm. Initially, the measured current values are identical, as can be observed by comparing the currents measured in stirred and quiescent solutions (steady current values at τ = 0 are shown in Fig. 7). The fact that the effect of bulk electrolyte properties are observed only in time follows from the observation that polarization curves recorded in different volumes of electrolyte were identical (Fig. 3).

Time-dependences of current density (calculated for the apparent surface area) at a potential of –1.1 V for electrodes with different surface areas: (1) 1, (2) 2, and (3) 5 cm2 in (1, 2, 3 ′) stagnant and (3′′) stirred solutions.
The facts that stirring has an effect on the measured quantity and that it increases with time are indicative of the mixed diffusion-kinetic nature of the measured current (it is kinetic, since the surface roughness, and therefore the true surface area, grow in time).
The tungsten content of coatings prepared by galvanostatic electrodeposition for 1.5 h is shown in Fig. 8. They were obtained in different electrolyte volumes (from 50 mL to 1 L), at different current densities (0.85–5 A/dm2), and using electrodes with different surface areas (0.5 to 8 cm2); and the tungsten content is plotted as a function of i/il, where il is the limiting current density (see also Fig. 4). In the unstirred electrolyte at 80°C, iI assumed a value of 2.3 A/dm2 (Fig. 4). As can be observed, the alloy tungsten content grows reaching almost 55 wt %, as both the current density and VCD increase (Fig. 8).

Alloy W content as a function of ratio of current density to limiting current density il at different VCD values.
The value for VCD determines the rate of change in the concentration of deposition-inducing metal species. Admittedly, according to Faraday law, it can be written as follows:
where I is the current, V is the electrolyte volume, E is the electrochemical equivalent, ɳ is the current efficiency, i is the current density, S is the electrodeposition surface area, ∆C is the concentration difference, and τ is the time.
In contrast, change in the concentration of deposition-inducing metal species is a function of time:
Equations (1) and (2) suggest that the bulk concentration of deposition-inducing metal species must decrease with increases in VCD and surface area and with time of plating.
Figure 9a shows electronic absorption spectra in the visible range for the initial electrolyte (1) and the electrolyte after 1.8 A h/L of use (2), which feature an absorption maximum at λ = 730 nm. As can be seen, the absorbance of electrolyte increased after prolong electroplating, which indicates an increase in the content of its low-molecular-weight fraction. Similar absorption spectra of the electrolyte are shown in Fig. 9b: before electrodeposition (1) and after 15 min of a potentiostatic electrodeposition at –1.1 V on electrodes with different surface areas ((2) 1 and (3) 5 cm2), as well as for the electrolyte used and then left for one day (4). It can be recognized that (i) the electrolyte composition changed during electrolysis, (ii) more pronounced changes are observed as the surface area increases, and (iii) the electrolyte behavior, in terms of visible spectra, recovers after several days.

Electronic absorption spectra of the electrolyte: (a) (1) initial and (2) with an extent of use (bath age) of 1.8 A h/L; (b) (1) before and after potentiostatic depositions at –1.1 V on working electrodes with a surface area of (2) 1 and (3) 5 cm2, as well as on the next day after electrodeposition (4).
It is to be noted that in this case the absorbance of solution increases rather than decreases during electrolysis. Since the total concentration remains essentially constant within a single deposition experiment, the increase in concentration of low-molecular-weight species is a result of a decrease in the concentration of the polymeric complex which recovers close to its initial level over time (Fig. 9b).
The increase in the low-molecular-weight fraction may result exclusively from the decrease in the concentration of deposition-inducing metal species, a high-molecular-weight complex, because it degrades into its constituents while performing its catalytic function. Further, the process of transformation between low- and high-molecular-weight products is slow.
Thus, the results obtained is further evidence for the fact that the deposition-inducing metal complex is a high-molecular-weight complex and acts as a catalyst for the tungsten reduction to form the alloy, which results in its degradation into low-molecular-weight products. The reverse transition is also observed and it depends on pH, as will be shown below; however, this process is extremely slow under the experimental conditions of this study.
Expressions (1) and (2) show that the change in the bulk concentration of deposition-inducing metal species must depend on both the electrodeposition surface area and electrodeposition time, and this was substantiated by the experimental results presented in Figs. 10 and 11. The observed nonmonotonic dependences of the alloy tungsten contents cannot be a result of experimental error, since this pattern was periodically observed in different experiments in both stirred and quiescent solutions and the values for the proportion of refractory component were measured sufficiently accurately (low uncertainty).

Effect of deposition time on the alloy tungsten content in electrodeposition at –1.1 V on electrodes with a surface area of (1) 1 and (2) 5 cm2 in both stirred and quiescent solutions.

Effect of deposition time on the alloy tungsten content in electrodepositions at –1.1 V from (1) the electrolyte with an extent of use of 1.8 A h/L 1 and (2) the initial electrolyte.
We note that the alloy tungsten content increases or varies nonmonotonically rather than decreases when the bulk concentration of deposition-inducing agent diminishes (Figs. 10 and 11). In addition, it grows with the bath age (Figs. 9a and 11). The conglomeration of presented results can be understood if we assume that there are several pathways for incorporation W into the alloy: (a) continuous generation (during the entire length of electrolysis) of the intermediate formed at stage I (Figs. 3 and 4) and (b) the reduction of Co–W complex at stage II formed in the solution. Moreover, the W : Co ratio in this complex is likely to shift toward higher proportion of W when depositing from more extensively used electrolyte. This supposition seems to be fairly reasonable, since the alloy Co content is greater than 70 at %; i.e., it is in this ratio that the alloying components make up the alloy, thus increasing the W concentration in solution and, therefore, in the coating. Results of comprehensive study of nonmonotonic variation in the alloying components will be reported in a later article.
Effect of the Side Reaction of Hydrogen Evolution on the Alloy Composition
From earlier studies [13, 30–33], a review [2], and our present experimental results, it can be concluded that the side reaction of hydrogen evolution has a considerable effect on the content of W (or any other refractory metal) in the alloy prepared by induced codeposition. Shown in Fig. 12 is the dependence of alloy tungsten content on the current efficiency in potentiostatic electrodeposition at –1.1 V on electrodes with different surface areas and in experiments with different deposition times (depositions on the 5 cm2 electrode were performed in both quiescent and stirred solutions). Interestingly, the data for tungsten content of alloys obtained under considerably different experimental conditions lie on a single (linear) trend. This is also reinforced by the results of galvanostatic depositions presented in Fig. 2, which show that the alloy tungsten content increases with decreasing the current efficiency, i.e., when the proportion of the side reaction of hydrogen evolution grows. Since the HER
leads to alkalization of the electrolyte near the electrode, it is reasonable to assume that it is alkalization which causes a rise in the refractory metal content of the alloy. This was also noted in previous studies [26, 27].

The alloy tungsten content as a function of current efficiency (CE) in potentiostatic electrodeposition at ‒1.1 V on electrodes with surface areas of (1) 1, (2) 2, and (3) 5 cm2 for different deposition durations.
At the same time, it must be taken into account that increases in pH cause polymerization of the deposition-inducing metal complex. For the cobalt– and iron–citrate complexes, the polymerization begins at pH > 5 [34] and pH > 4 [29], respectively. The polymerization of iron–citrate complex can be represented by the reaction [29]
On the other hand, separation of cobalt–citrate complexes by gel filtration demonstrated that alloy deposition occurs only when a high-molecular-weight component is present in the electrolyte [25], suggesting that the deposition-inducing metal is the central ion of the citrate complex.
The results of the present study provide answers to the question concerning the contribution of reactions (1) and (2) to the MSE in general, and the effect of VCD on the refractory metal content of the alloy prepared by induced codeposition (under galvanostatic conditions), in particular. Since the concentration of polymeric complex is considerably lower than the total solution concentration of deposition-inducing metal, the high rate of change of the former (i.e., at high IV) causes a diminution of the limiting current density iI, because it is controlled by the bulk concentration of polymeric complex, while the potential shifts cathodically (Fig. 2) resulting in an increase in the proportion of the side reaction, a decrease in the current efficiency of the target process, and an increase in the proportion of reaction (1). This further leads to a local pH rise, the shift in equilibrium toward the formation of polymeric products (similar to reaction (2)), and, therefore, the increase in the alloy tungsten content (Fig. 12), because the alloy is formed only in the presence of the high-molecular-weight component in the electrolyte, as previous studies showed [25].
It can also be concluded that the significance of reaction (I) is not in the fact that hydrogen reduces intermediates of the iron group metal and the refractory metal, rather it contributes to a local change in pH, which ensures the formation of polymeric citrate complex of deposition-inducing metal, which acts as the catalyst.
It is remarkable that the resulting nanostructured coating (at high alloy tungsten contents) consists of series of lamellae of a chain polymer, described in [35] (section 2.1.8), with a size of ~8 nm (Fig. 13). Such structures are formed during crystallization of polymers from solutions [35], which further highlights that the polymeric identity of the complex of deposition-inducing agent is a critical feature of alloy formation by induced codeposition of iron group metals with tungsten.

Co–W nanostructure deposited from a gluconate bath [36].
SINGLE NATURE OF THE MACROSCOPIC SIZE EFFECT IN VARIOUS FORMS OF ITS MANIFESTATION
The MSE (i.e., the dependence of composition and properties of coatings prepared by induced codeposition on the electrodeposition surface area) has various manifestations, which means that it can be of different types (or forms) (Figs. 1 and 2). First, it manifests as an alteration of the composition and microhardness of the surface layer (Fig. 1). It can also manifest as a change in the bulk composition of deposited coating (Fig. 2). In this case, the VCD or the surface area (for a definite surface current density and electrolyte volume) have different effects on the composition and, therefore, properties of deposited alloy. In the first case, an increase in current density results in diminution of microhardness (Fig. 1), while in the second case it leads to an increase in the alloy tungsten content (Fig. 2) and, therefore, an increase in microhardness.
As was shown earlier [21], the first form of the considered effect is associated with specific features of nanonucleation, namely, the interaction between the intermediate species and solvent (water) molecules under specified reaction conditions (Fig. 14). The schematics presented in Fig. 14 describes the final stage of the overall process. The formation of intermediates occurs at earlier stages. A tungsten cluster (the term used in [7]) is formed at stage I (Fig. 3) and is an intermediate of the reduction of tungsten species, and one possibility of this cluster (so-called tungsten blue) is arbitrarily shown in Fig. 14. The following events take place when a potential of –0.76 V is reached: a) the cobalt–citrate complex undergoes reduction giving rise to an intermediate, (so-called “iron cluster”) [7] that reacts with the tungsten intermediate (the tungsten cluster) at the electrode surface; and (b) reduction of the Co–W–citrate complex. The surface coverage of processes just described determines the ratio of alloying components by controlling the alloy tungsten content. At a high rate of diminution of the deposition-inducing species and/or of the Co–W–citrate complex (i.e., at high Iv), however, another possibility arises, which leads to the involvement of solvent (water) into the reaction (Fig. 14). This results in the formation of oxy-hydroxide surface layer and hydrogenation of the metal phase, and, consequently, a decrease in the measured microhardness. It is readily apparent that this process is a consequence of the decrease in polymer complex concentration on the surface because of an increase in VCD (see Eq. (1)).

Schematic presentation of the formation of alloy and of surface layers.
At the same time, a decrease in the bulk concentration of deposition-inducing metal species, while maintaining the current density constant, results in a cathodic shift in potential, the involvement of reaction (1), a local pH rise, promotion of polymerization of deposition-inducing metal complex, and consequently, an increase in the alloy tungsten content (Fig. 2). As previous studies showed in the context of electrodeposition of Fe–W alloys from a citrate–glycolate bath, an increase in the alloy tungsten content leads to alteration of the alloy structure due to the formation of intermetallics, along with solid solutions [37]. In addition, it is the decrease in the surface concentration of deposition-inducing metal complex that leads to a decrease in the nucleus size during the phase formation, which further leads to the formation of nanocrystalline surface thereby increasing the microhardness [21]. An increase in the coating tungsten content involves the progression from a crystalline to nanocrystalline structure ([35, 38]). Put differently, the formation of nanocrystalline coatings by induced codeposition of the iron group metals with refractory metals is a consequence of the deposition-inducing metal species having a polymeric form. Clearly, the polymeric form of the complex also explains why specific features of nanonucleation in this electrolytes can be placed in the corresponding theoretical framework by assuming that the electrolytes display fractal properties [21]. At the same time, it must be taken into account that the coating tungsten content depends not only on VCD but also the (surface) current density i (Fig. 8).
CONCLUSIONS
(1) The insights of the present work indicate that the polymeric identity of the deposition-inducing metal complex, which is a cobalt–citrate complex (in the context of considered experiments) with a linear structure and a molecular weight of ~2 × 105 Da, is its critical feature.
(2) The reduction of this complex leads to deposition of the alloy with a nanocrystalline structure, which consists of lamellae with an average size of ~8 nm elongated perpendicular to the surface.
(3) The formation of such a structure takes place as a result of the reaction between the deposition-inducing species and an intermediate of the reduction of the tungsten species, which is a high-molecular-weight tungsten complex formed at potentials more anodic than the reduction potential of the Co–W complex. The fact that it also has a high molecular weight is indicated by the results of electrochemical impedance study, which showed that the charge separation region undergoes tangential stresses within the hydrodynamic boundary layer.
(4) When the overall concentration of the deposition-inducing metal complex is constant due to the formation of the high-molecular-weight polymer, the decrease in its molar concentration results in high current loading on the bath, which causes the macroscopic size effect in the composition and properties of resulting alloys (i.e., the effect of electrodeposition surface area on the deposit composition and properties).
(5) The observed macroscopic size effects can be of different types: a) alterations of surface properties due to the formation of oxy-hydroxide layers and hydrogenation and b) effect of volume current density on the ratio of alloy contents of W and an iron group metal.
(6) Apart from controlling deposition parameters conventional to any process in electrochemical materials science, one basic requirement (condition) to be considered in scaling up of electrodeposition technologies of coatings of the iron group metals with tungsten is the maintenance of current loading (i.e., volume current density) on the bath at a constant level.
REFERENCES
Eliaz, N. and Gileadi, E., Induced codeposition of tungsten, molybdenum and rhenium with transition metals, Modern Aspects Electrochem., 2008, vol. 42, p. 191. https://doi.org/10.1007/978-0-387-49489-0_4
Tsyntsaru, N., Cesiulis, H., Donten, M., Sort, J., et al., Modern trends in tungsten alloys electrodeposition with iron-group metals, Surf. Eng. Appl. Electrochem., 2012, vol. 48, no. 6, p. 491. https://doi.org/10.3103/S1068375512060038
Yapontseva, Yu.S., Dikusar, A.I. and Kyblanovskii, V.S., Study of the composition, corrosion and catalytic properties of Co–W alloys electrodeposited from a citrate pyrophosphate electrolyte, Surf. Eng. Appl. Electrochem., 2014, vol. 50, p. 330. https://doi.org/10.3103/S1068375514040139
Cesiulis, H., Tsyntsaru, N., Podlaha, E.J., Li, D., et al., Electrodeposition of iron-group alloys into nanostructured oxide membranes: Synthetic challenges and properties, CNANO, 2018, vol. 15, p. 84. https://doi.org/10.2174/1573413714666180410154104
Maliar, T., Cesiulis, H., and Podlaha, E.J., Coupled electrodeposition of Fe-Co-W alloys thin films and nanowires, Front. Chem., 2019, vol. 7, p. 542. https://doi.org/10.3389/fchem.2019.00542
Krasikov, A.V. and Krasikov, V.L., Mechanism of electrodeposition of nickel–tungsten alloy from a pyrophosphate electrolyte, Bull. SPbSTI (TU), 2016, vol. 36 (62), p. 12. https://doi.org/10.15217/issn1998984-9.2016.36.12
Krasikov, A.V. and Krasikov, V.L., Mechanism of induced codeposition of alloys and some single refractory metals, Bull. SPbSTI (TU), 2016, vol. 37, p. 8. https://doi.org/10.15217/issn1998984-9.2016.37.8
Brenner, A., Electrodeposition of Alloys. Principles and Practice, New York: Academic, 1963. https://doi.org/10.1016/C2013-0-07892-9
Belevskii, S.S., Yushchenko, S.P. and Dikusar, A.I., Anomalous electrodeposition of Co–W coatings from citrate electrolyte due to the formation multinuclear heterometalic complexes in the solution, Surf. Eng. Appl. Electrochem., 2012, vol. 48, no. 1, p. 97. https://doi.org/10.3103/S1068375512010036
Podlaha, E.J. and Landolt, D., Induced codeposition. I. Experimental investigation of Ni–Mo alloys, J. Electrochem. Soc., 1996, vol. 143, p. 885. https://doi.org/10.1149/1.1836553
Podlaha, E.J. and Landolt, D., Induced codeposition. II. Mathematic modeling of Ni–Mo alloys, J. Electrochem. Soc., 1996, vol. 143, p. 893. https://doi.org/10.1149/1.1836554
Podlaha, E.J. and Landolt, D., Induced codeposition. III. Molybdenum alloys with nickel, cobalt and iron, J. Electrochem. Soc., 1997, vol. 144, p. 1672. https://doi.org/10.1149/1.1837658
Holt, M.L. and Vaaler, L.E., Electrolytic reduction of aqueous tungsten solutions, J. Electrochem. Soc., 1948, vol. 94, p. 50. https://doi.org/10.1149/1.2773824
Fukushima, H., Akiyama, T. and Akagi, S., Role of iron group metals in the induced codeposition of molybdenum from aqueous solution, Trans. Jpn. Inst. Metal., 1979, vol. 20, p. 358. https://doi.org/10.2320/matertrans1960.20.358
Sun, S. and Podlaha, E.J., Electrodeposition of Mo-rich MoNi alloys from an aqueous electrolyte, J. Electrochem. Soc., 2011, vol. 159, p. D1. https://doi.org/10.1149/2.091202jes
Silkin, S.A., Gotelyak, A.V., Tsyntsaru, N.I. and Dikusar, A.I., Size effect of microhardness of nanocrystalline Co–W coatings produced from citrate and gluconate solutions, Surf. Eng. Appl. Electrochem., 2015, vol. 51, no. 2, p. 228. https://doi.org/10.3103/S106837551503014X
Gotelyak, A.V., Silkin, S.A., Yahova, E.A. and Dikusar, A.I., Effect of pH and volume current density on deposition rate and microhardness of Co–W coatings electrodeposited from concentrated boron-gluconate electrolyte, Russ. J. Appl. Chem., 2017, vol. 90, no. 4, p. 541. https://doi.org/10.1134/S1070427217040085
Silkin, S.A., Gotekyak, A.V., Tsynysaru, N.I. and Dikusar, A.I., Electrodeposition of alloys of the iron-group metals with tungsten from citrate and gluconate solutions. Size effect of microhardness, Surf. Eng. Appl. Electrochem., 2017, vol. 53, no. 1, p. 7. https://doi.org/10.3103/S1068375517010136
Belevskii, S.S., Gotelyak, A.V., Silkin, S.A. and Dikusar, A.I., Macroscopic size effect of the microhardness of electrodeposited iron-group metal–tungsten alloy coatings. Impact of electrode potential and oxygen-containing impurities, Surf. Eng. Appl. Electrochem., 2019, vol. 55, no. 1, p. 46. https://doi.org/10.3103/S1068375519010058
Belevskii, S.S., Danilchuk, V.V., Gotelyak, A.V., Lelis, M., et al., Electrodeposition of Fe–W coatings from citrate bath. Impact of anode material, Surf. Eng. Appl. Electrochem., 2020, vol. 56, no. 1, p. 1. https://doi.org/10.3103/S1068375520010020
Baranov, S.A. and Dikusar, A.I., Kinetics of electrochemical nanonucleation during induced codeposition of iron group metals with refractory metals (W, Mo, Re), Surf. Eng. Appl. Electrochem., 2022, vol. 58, no. 5, p. 429. https://doi.org/10.3103/S1068375522050027
Myrzak, V., Gotelyak, A.V. and Dikusar, A.I., Size effects in the surface properties of electrodeposited alloys between iron-group metals and tungsten, Surf. Eng. Appl. Electrochem., 2021, vol. 57, no. 4, p. 409. https://doi.org/10.3103/S1068375521040128
Belevskii, S.S., Gotelyak, A.V., Ivashku, S.Kh., Kovalenko, K.V., and Dikusar, A.I., Anodic dissolution of surface layers as a means of increasing the microhardness of alloy coatings of iron group metals with tungsten prepared by induced codeposition, Surf. Eng. Appl. Electrochem., 2023, vol. 59, pp. 549–555.
Belevskii, S.S., Buravets, V.A., Yushchenko, S.P., Zgardan I.M., et al., Gel-chromatographic separation of boron-gluconate electrolyte for obtaining nano-crystalling Co–W coatings. Part I. Composition and electrochemical activity of components, Surf. Eng. App-l. Electrochem., 2016, vol. 52, no. 4, p. 350. https://doi.org/10.3103/S1068375516040049
Belevskii, S.S., Buravets, V.A., Yushchenko, S.P., and Dikusar, A.I., Gel-chromatographic separation of boron-gluconate electrolyte for obtaining nano-crystalling Co–W coatings. Part II. Electrochemical activity of separation products and their role in process manufacturing alloy, Surf. Eng. Appl. Electrochem., 2016, vol. 52, no. 5, p. 420. https://doi.org/10.3103/S1068375516050057
Belevskii, S., Silkin, S., Tsyntsaru, N., Cesiulis, H. and Dikusar, A., The influence of sodium tungstate concentrations on the electrode reactions at iron–tungsten alloy electrodeposition, Coatings, 2021, vol. 11, no. 8, 981-1. https://doi.org/10.3390/coatings11080981
Belevskii, S.S., Cesiulis, H., Tsyntsaru, N.I. and Dikusar, A.I., The role of mass transfer in the formation of the composition and structure of CoW coatings electrodeposited from citrate solutions, Surf. Eng. Appl. Electrochem., 2010, vol. 46, no. 6, p. 570. https://doi.org/10.3103/S1068375510060050
Spiro, T.G., Pape, L., and Saltman, P., The hydrolytic polymerization of ferric citrate I. The chemistry of polymer, JACS, 1967, vol. 89, p. 5555. https://doi.org/10.1021/ja00998a008
Spiro, T.G., Bates, G., and Saltman, P., The hydrolytic polymerization of ferric citrate II. The influence of excess citrate, JACS, 1967, vol. 89, p. 5559. https://doi.org/10.1021/ja00998a009
Clark, E. and Leitzke, H.H., The mechanism of the tungsten alloy plating process, J. Electrochem. Soc., 1952, vol. 99, p. 245. https://doi.org/10.1149/1.2779712
Ernst, D.W., Amile, R.F. and Holt, M.L.J. Electrodeposition of molybdenum alloys from aqueous solutions, J. Electrochem. Soc., 1955, vol. 102, 461. https://doi.org/10.1149/1.2430124
Ernst, D.W. and Holt, M.L., Cathode potentials during the electrodeposition of molybdenum alloys from aqueous solutions, J. Electrochem. Soc., 1958, vol. 105, p. 686. https://doi.org/10.1149/1.2428691
Oue, S., Nakano, U., Kobayashi, S. and Fukushima, H., Structure and codeposition behavior of Ni-W alloys electrodeposited from ammoniacal citrate solutions, J. Electrochem. Soc., 2009, vol. 156, p. D17. https://doi.org/10.1149/1.3006389
Belevskii, S.S., Yuchchenko, S.P. and Dikusar, A.I., Electrodeposition of nanocrystalline Co–W coatings from citrate electrolytes under conditions of controlled hydrodynamics. I. Co electrodeposition, Surf. Eng. Appl. Electrochem., 2009, vol. 45, no. 6, p. 446. https://doi.org/10.3103/S1068375509060027
Gleiter, H., Nanostructured materials: basic concepts and microstructure, Acta Materiala, 2000, vol. 48, no. 1, p. 1. https://doi.org/10.1016/S1359-6454(99)00285-2
Weston, D.P., Gill, S.P.A., Fay, M., Harris, S.J., et al., Nanostructure of Co–W alloy electrodeposited from gluconate base, Surf. Coat. Technol., 2013, vol. 236, p. 75. https://doi.org/10.1016/j.surfcoat.2013.09.031
Nicolenco, A., Tsyntsaru, N., Fornell J., Pellicer, E., et al., Mapping of magnetic and mechanical properties of Fe–W alloys electrodeposited from Fe (III)-based glycolate-citrate bath, Mater. Design, 2018, vol. 139, p. 429. https://doi.org/10.1016/j.matdes.2017.11.011
Nicolenco, A., Tsyntsaru, N. and Cesiulis, H., Fe(III)-based ammonia-free bath for electrodeposition of Fe–W alloys, J. Electrochem. Soc., 2017, vol. 164, no. 9, p. D590. https://doi.org/10.1149/2.1001709jes
Funding
The study was supported within the H2020 program; the Marie Skłodowska-Curie Actions Innovation Program RISE-2017 (project no. 778357); by the National Agency for Research and Development, Moldova within the project “Manufacturing of New Micro- and Nanostructuring Materials by Physico-Chemical Methods and the Elaboration on Their Base” (project no. 20.8009.5007.18); and within the budget funding of the Shevchenko Pridnestrov’e State University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note.
Allerton Press remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Dikusar, A.I., Belevskii, S.S. Macroscopic Size Effect in the Composition and Properties of Alloys of Iron Group Metals with Tungsten Prepared by Induced Codeposition: Alloy Deposition Mechanism and Its Implications. Surf. Engin. Appl.Electrochem. 59, 699–711 (2023). https://doi.org/10.3103/S106837552306008X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3103/S106837552306008X