
Abstract
Gene targeting is extensively used to generate designer mouse mutants and to study gene function in vivo. Knockout mice that harbor a null allele in their germline provide appropriate genetic models of inherited diseases and often exhibit embryonic or early postnatal lethality. To study gene function in adult mice and in selected cell types, a refined strategy for conditional gene inactivation has been developed that relies on the DNA recombinase Cre and its recognition (loxP) sites. This process has traditionally relied on the complex process involving genome editing in embryonic stem (ES) cells despite its limitations, including incorrect targeting or cassette structure, and difficulties with germline transmission of the allele from chimeric mice. CRISPR-Cas9 gene editing technology has considerably facilitated the generation of mouse knockout alleles, relieving many of the cumbersome and time-consuming steps of traditional mouse ES cell technology. However, the generation of conditional knockout alleles remains an important challenge. An earlier study reported up to 16% efficiency in generating conditional knockout alleles in mice using 2 single guide RNAs (sgRNA) and 2 single-stranded oligonucleotides (ssODN), which has been questioned by another report combining data from multiple transgenic cores. With the advent of CRISPR/Cas9 as a mouse genome modification tool, we assessed the efficiency of using this method in creating conditional targeted alleles in three genes, phosphatase and actin regulator 1 (Phactr1), apolipoprotein A-I (ApoA1), and actin-related protein T2 (Actrt2). Even though overall success rate was low—about 2.5%—we show that it’s possible to reliably generate conditional knockout alleles using CRISPR/Cas9 on a consistent basis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Targeted gene mutation in the mouse is a primary strategy to understand gene function and relation to phenotype. However, according to the International Mouse Phenotyping Consortium (http://www.mousephenotype.org/), more than 60% (284/459) of knockout mouse strains (C57BL/6N background) show a prenatal lethality phenotype. To overcome this limitation and study the gene functions in adult mice, a refined knockout strategy termed conditional mutagenesis has been developed that enables to inactivate a target gene only in a selected cell type [1, 2]. The most commonly-used system for conditional knockout is the cell type-specific expression of the DNA recombinase Cre, a site-specific DNA recombinase derived from the PI phage, while the target gene is modified by the insertion of 34-bp recombinase recognition (loxP) sequences [2, 3]. The loxP recognition site for Cre is composed of two 13-bp inverted repeats separated by an 8-bp asymmetric spacer that determines the orientation of the loxP site [4]. Cre-mediated recombination between two loxP sites results in the reciprocal exchange of DNA strands between these sites. Depending on the orientation and location of the two sequences, different products are obtained. In this system called Cre/lox, a region of interest flanked by two lox sites (floxed) positioned in the same orientation, is deleted or inverted by Cre-mediated recombination, leading to gene knockout only in a Cre-expressing cell. As Cre does not require cofactors or accessory proteins to mediate loxP-specific DNA recombination and exhibits optimal activity at 37°C, the Cre/loxP system is the most widely used recombination tool in ES cells and mice [5]. Today, more than 1.300 strains of Cre mice that show tissue- and stage-specific expression of recombinases are available from bio-resource repositories in several countries (International Mouse Strain Resource; http://www.findmice.org/index). By contrast, researchers have to produce a mouse with a floxed allele in a gene of interest in many cases.
Traditionally, flox mice have been obtained by gene targeting in embryonic stem cells followed by production of germline chimeric mice. However, generating precise modifications in endogenous genes is very complicated. In addition, it takes about a year or more to obtain flox mice by production of chimeric mice and mating of their offspring. Recently, genome editing using direct injection of engineered endonucleases or RNA-guided nucleases into zygotes has greatly accelerated the production of gene-modified animals. The most popular system, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas), is based on RNA-guided nucleases. The minimal system consists of the Cas9 endonuclease and a target-specific guide RNA (gRNA) [6]. In human cells, Cas9 and gRNA can induce DNA double-strand breaks (DSBs) at target sequences, leading to targeted mutations by non-homologous end joining (NHEJ) [7–10]. Furthermore, direct injection of these components into zygotes generates NHEJ-mediated mutant mice [11–13]. Later it was determined that Cas endonucleases can generate a precise double strand break (DSB) in the DNA under a chimeric single guide RNA (sgRNA) [6]. In an earlier study, a high success rate (16%) of targeting LoxP sites in cis was reported by using 2 sgRNAs and 2 single-stranded oligonucleotides (ssODN) containing LoxP sites (2sgRNA-2ssODN) flanking a targeted critical exon [14]. Nevertheless, the recent study analyzing combined reports from 17 transgenic core facilities shows that results of this approach are inconsistent [15].
Since this method could be a powerful tool to generate flox mice because it is not necessary to construct a knock-in vector via a complicated process, and flox mice can be obtained in a short period of time (e.g., in a month), the goal of our study was to investigate the efficiency of the 2sgRNA-2ssODN method for the generation of several conditional knockout alleles.
MATERIAL AND METHODS
SgRNA Design
Three single guide RNAs (sgRNAs) were designed using proprietary algorithm by CRISPR Core Partnership Program by Sigma Millipore (Merck KGaA, Darmstadt, Germany) per each LoxP insertion site (Table 1 shows guide RNA sequences originally selected for our first target gene Phactr1). These guide RNAs were then validated via CEL I mutation detection assay (also by Sigma Millipore) and the guide showing the highest activity was chosen for each target site for injections and also to design and synthesize matching single stranded DNA oligo donor (ssDNA, also produced by Sigma Millipore). Cas9 mRNA was also provided by Sigma Millipore (Cat #CAS9MRNA). Concentrations and site of injection for Cas9 mRNA, sgRNA, and template repairs for each locus are indicated in Table 2. Cas9 mRNA, sgRNAs, and oligonucleotides were mixed immediately prior to injection in ТЕ buffer, and centrifuged for 10 min at 14 000 rpm.
Mouse Strains and Husbandry
B6.Cg-Tg(Sox2-cre)1Amc/J (Sox2-Cre, stock 8454) mice were purchased from the Jackson Laboratory (Bar Harbor, ME); Crl:CD1(ICR) and C57BL/6NCrl (C57BL/6N) mice were purchased from Charles River (Wilmington, MA). Mice were housed in the AAALAC-accredited barrier animal facility at TIGM, and were maintained under 12/12 h light cycle with food and water provided ad libitum. All studies were reviewed and approved by the Institutional Animal Care and Use Committee of the Texas A&M University, and followed National Institutes of Health (NIH) guidelines for the Care and Use of Laboratory Animals.
Mouse Zygote Microinjection
Three to five week-old C57BL/6N females were superovulated by intraperitoneal injection of Pregnant Mare Serum Gonadotropin (5IU, ProSpec-Tany TechnoGene Ltd., Israel) followed by intraperitoneal injection of Human Chorionic Gonadotropin hormone (5IU, Sigma-Aldrich, Inc.) 48 h later. Superovulated females were mated with 8 to 20 week-old stud males. The mated females were euthanized next day and the zygotes were collected from their oviducts in microinjection medium (5% FBS added to HEPES-buffered D-MEM (ThermoFisher Scientific, Waltham, MA)). Injections were performed under an inverted microscope, associated micromanipulator, and the microinjection apparatus, Eppendorf Transjector 5246, with in-house pulled glass capillaries. Fertilized oocytes were injected into either the pronuclei or cytoplasm with the prepared CRISPR/Cas9 reagents using air-regulated compensation and an injection pressure of 90–115 psi in order to create a continuous flow of reagents [16]. Due to the continuous flow of reagents, fertilized oocytes with injections into the pronuclei received CRISPR/Cas9 reagents in both pronuclear and cytoplasmic regions. Injected zygotes were incubated at 37°C, 5% CO2 until transplantation. Approximately nine zygotes were surgically transferred into each oviduct of the pseudo-pregnant ICR recipient females or cultured overnight at 37°C and then transferred into recipients at the 2-cell stage of development.
Genotyping
Mice were screened prior to weaning at the age of 12–17 days. For genotyping, tail snip DNA was extracted using tail lysis procedure with Proteinase K. The lysis (L) buffer contained 25 mM Tris-HCl pH 7.5, 100 mM NaCl, 50 mM EDTA, 1% Sarkosyl, 5 mM DTT, 0.05 mM Spermidine and 2 μl proteinase K (Sigma) per 100 μl of the buffer. Mouse tails in buffer L + proteinase K (300–400 μL/tail) were incubated overnight at 65°C, diluted 1 : 40, heated at 95°C for 5 min and used for PCR reactions. Primers were designed to amplify the regions encompassing the integrated LoxP sequence (Fig. 1). PCR was performed using LongAmpTM Taq Master Mix (New England Biolabs) under standard PCR conditions. Primer Sequences (5' to 3') and expected PCR bands for the Phactr1 knockout allele were as follows:

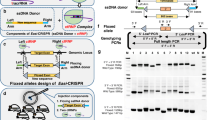
Schematic process for creation of Phactrl conditional knockout allele. (A) Wild type locus showing exons 7, 8 and 9; exon 8 is chosen as a target for inserting LoxP sites. gRNAs target introns 7 and 8. (B) CRISPR components—sgRNA, ssODN donors and a Cas9 source—delivered into one-cell stage zygotes via microinjection. (C) The conditional (“floxed”) knockout allele showing target exon 8 with flanking LoxP sites. (D) Excision of the target exon 8 following exposure to Cre recombinase (“flox del” locus). Genomic locations of the genotyping primers (F, R, upF1, dwF1, upR1 and dwR1) are also shown.
Upstream LoxP PCR: upF1 + upR1, 208 bp
Phactr1 upF1 GCT CAC CCC CAA TAA CTT CGT ATA
Phactr1 upR1 GTCCAACTCACCTCCACTGA
Downstream LoxP PCR: dwF1 + dwR1, 216 bp
Phactr1 dwF1 GGC AAG TTC С AG GTC AGT GA
Phactr1 dwR1 GGCCTGAATTCATAACTTCGTATAGC
Upstream LoxP to Downstream LoxP PCR: upF1 + dwR1, 1572 bp
PCR confirming the floxed fragment excision: F+R, 2063 bp (wt)/2144 bp (floxed) + 557 bp (flox del)
Phactr1 F GCAGCCTTCCTGCTGCAGCTTTCT
Phactr1 R GCTTTGCTTCCAGTACCCTGATCT
Similar strategies were designed for the other two alleles, ApoA1 and Actrt2. The PCR products were then purified with a PCR Clean-Up System kit according to the manufacturer’s instructions. Sanger sequencing was performed in Texas A&M Sequencing Core facility. The entire regions encompassing both the guide cleavage sites were amplified to assess for integrity of the LoxP sites and presence of unintended modifications between the cleavage sites.
RESULTS
We report the construction and preliminary analysis of several conditional knockout mouse strains (Phactr1, ApoA1, Actrt2). All three alleles were generated using standard procedures following the same scheme described for Phactr1 (Fig. 1). The first step of the process was design of sgRNAs and their validation. Three guide RNAs were selected per each target site based on their uniqueness and specificity, of which only one was chosen for further work based on results of the Cel I validation assay (Table 1). Only then the ssDNA donors would be designed and synthesized following several basic principles: 70 nt for each homology arm and LoxP cassette inserted between guide RNA sequence and Protospacer Adjacent Motif (РАМ) (Fig. 2A and Table 2).

Creation of Phactrl conditional knockout allele. (A) sgRNA and ssODN donor sequences for inserting LoxP sites. (B) Microinjection results. (C) Genotyping confirmation of the correct LoxP sites insertions. (D) Genotyping confirmation that the LoxP sites are inserted in cis-orientation (same strand). (E) Genotyping confirmation of the floxed fragment excision following exposure to Cre recombinase.
Injections were performed according to standard methods with the aim to deliver material both inside the nucleus and cytoplasm. The number of surviving embryos varied from experiment to experiment, but would normally stay in 55–75% range and the embryo to live offspring conversion rate was in the range of 12–22% (Table 3).
Original genotyping PCR screen would identify single LoxP insertions followed by long—distance PCR to confirm cis-orientation of the LoxP sites on the chromosome. As expected, most of the founders contained single LoxP insertions as well as NHEJ-based deletions between guide RNA targets, and only a few mice carried correct insertions at both sites. Interestingly, all 6 founders with both LoxP site insertions produced for these 3 projects carried them in cis-orientation, which contradicts recent reports of regular insertions in trans [15]. Further sequencing of the genomic DNA surrounding LoxP site insertions revealed no unintended mutations in the founders, also contrasting recent reports of finding numerous mutations at those sites by other labs [15]. The overall efficiency of correct editing events occurring simultaneously at both sites was approximately 2.5% (Fig. 2B, Table 3).
Founder males or females were bred to C57BL/6 females or males for germline transmission of the mutant alleles. A germline transmission event was scored when any F0 founder produced progeny with the correct genomic locations of the LoxP sites. The progeny were analyzed for transmission of the mutation using site-specific PCR protocols (Figs. 2C and 2D). The Fl progeny heterozygous mutants revealed no visible phenotype. To continue with the functional characterization of these tissue-specific knockouts we obtained homozygous (flox/flox) ApoA1 and Actrt2 alleles and started breeding with tissue-specific Cre strains. Correct removal of the targeted genomic fragment was confirmed by appropriate genotyping PCR (Fig. 2E shows results of the Cre-mediated deletion for Phactr1 following breeding with Sox2-Cre line). Homozygous mice were produced at the expected Mendelian rate and didn’t display any gross abnormalities, further suggesting that these knockouts do not carry any unwanted genome modifications and can be used as intended.
DISCUSSION
CRISPR-Cas9 technology has greatly facilitated the generation of mouse lines containing knockout or knock-in alleles. Over the past several years TIGM has created 20 mouse strains carrying various site-specific genome modifications using this approach. However, the generation of conditional alleles remains a challenge using traditional ES cells and CRISPR-Cas9 gene editing technologies. An earlier paper demonstrated success approaching 16% efficiency with 2 chimeric sgRNAs and 2 single-stranded oligonucleotides donor approach to produce conditional alleles in mouse [14].
To evaluate the efficiency of the method, we performed experiments on production of 3 different conditional alleles. Although we observed numerous single LoxP site insertions and indels at the cleavage sites, the method was also successful in generating two LoxP sites in cis for all three alleles (Table 3). We noted the efficiency to insert the two LoxP sites simultaneously to be the best predictor of the likelihood of success in this approach. In fact, we haven’t found any trans locations whenever both LoxP site insertions were observed so far. We also noted a low success rate overall (~2.5%, representing the number of correctly edited mice to the total number of live offspring born for a particular project) to generate a conditional allele (Table 3). These results are comparable with previous reports demonstrating an important disparity in success rate varying from 0 to 7% of mice harboring two LoxP sites insertions in cis by microinjection [17–20]. We also have noted a number of deletions at the target sites following DNA cleavage [19]. Another possibility to improve the efficiency of the method is to avoid recombination between the target sites by placing the LoxP sites over hundreds of kb apart. This was reported previously for a success rate varying from 0 to 18% for 6 loci [21]. A recent report found the successful use of sequential introduction of the LoxP sites to improve its efficiency as much as 3 to 10 fold and avoid recombination between alleles [17], though it should be noted that such an approach requires a more protracted period of time to completion. Additional work has demonstrated over 5 fold improvement in targeting using a long ssODN [20, 22].
In conclusion, even though overall success rate is low—about an average of 300 zygotes were needed to generate 1 correctly targeted animal – it is possible to generate floxed alleles using CRISPR-Cas9 gene editing technology with 2sgRNA-2ssODN. We believe that Phactr1, ApoA1, Actrt2 conditional KO mice strains will serve as a valuable tool for researchers studying appropriate genetic models.
REFERENCES
Gu, H., Marth, J.D., Orban, P.C., Mossmann, H., and Rajewsky, K., Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting, Science, 1994, vol. 265, pp. 103–106.
Rajewsky, K., Gu, H., Kuhn, R., Betz, U.A., Muller, W., Roes, J., and Schwenk, F., Conditional gene targeting, J. Clin. Invest., 1996, vol. 98, pp. 600–603.
Sauer, B. and Henderson, N., Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P, Proc. Natl. Acad. Sci. U. S. A., 1988, vol. 85, pp. 5166–5170.
Hoess, R.H. and Abremski, K., Interaction of the bacteriophage P1 recombinase Cre with the recombining site loxP, Proc. Natl. Acad. Sci. U. S. A., 1984, vol. 81, pp. 1026–1029.
Brand,a, C.S. and Dymecki, S.M., Talking about a revolution: the impact of site-specific recombinases on genetic analyses in mice, Dev. Cell, 2004, vol. 6, pp. 7–28.
Jinek, M., et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity, Science, 2012, vol. 337, pp. 816–821.
Cho, S.W., Kim, S., Kim, J.M., and Kim, J.S., Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease, Nat. Biotechnol., 2013, vol. 31, pp. 230–232.
Cong, L., et al., Multiplex genome engineering using CRISPR/Cas systems, Science, 2013, vol. 339, pp. 819–823.
Jinek, M., et al., RNA-programmed genome editing in human cells, Elife, 2013, vol. 2. e00471. https://doi.org/10.7554/eLife.00471
Mali, P. et al. RNA-guided human genome engineering via Cas9, Science, 2013, vol. 339, pp. 823–826.
Mashiko, D. et al. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA, Sci. Rep., 2013, vol. 3355, no. 3. https://doi.org/10.1038/srep03355
Shen, B. et al. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting, Cell Res., 2013, vol. 23, pp. 720–723.
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering, Cell, 2013, vol. 153, pp. 910–918.
Yang, H., et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering, Cell, 2013, vol. 154, no. 6, pp. 1370–1379.
Gurumurthy, C., et al. Re-evaluating one-step generation of mice carrying conditional alleles by CRISPR-Cas9-mediated genome editing technology, bioRxiv, 2018, vol. 393231. https://doi.org/10.1101/393231
Behringer R., et al., Manipulating the Mouse Embryo: A Laboratory Manual, Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, 2014.
Horii, T., et al., Efficient generation of conditional knockout mice via sequential introduction of lox sites, Sci Rep., 2017, vol. 7, no. 1, p. 7891.
Bishop, K.A., et al., CRISPR/Cas9-mediated insertion of loxP sites in the mouse dock7 gene provides an effective alternative to use of targeted embryonic stem cells, G3 (Bethesda), 2016, vol. 6, no. 7, pp. 2051–2061.
Kueh, A.J., et al., An update on using CRISPR/Cas9 in the one-cell stage mouse embryo for generating complex mutant alleles, Cell Death Differ., 2017, vol. 24, no. 10, pp. 1821–1822.
Lanza, D.G., et al., Comparative analysis of single-stranded DNA donors to generate conditional null mouse alleles, BMC Biol., 2018, vol. 16, no. 1, p. 69.
Pritchard, C.E.J., Kroese, L.J., and Huijbers, I.J., Direct Generation of Conditional Alleles Using CRISPR/ Cas9 in Mouse Zygotes. Methods Mol Biol., 2017, vol. 1642, pp. 21–35.
Miyasaka, Y., et al., CLICK: one-step generation of conditional knockout mice, BMC Genomics, 2018, vol. 19, no. 1, p. 318.
ACKNOWLEDGMENTS
We would like to thank Dr. Christopher Lemke and Dr. Nicola van der Walt of CRISPR Core Partnership Program (Sigma Millipore/Merck KGaA, Darmstadt, Germany) for help with design and validation of CRISPR/cas9 materials.
Funding
This work was supported by Texas AgriLife Research (SS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest or financial disclosure statements. This article does not contain any studies involving animals or human participants performed by any of the authors.
About this article

Cite this article
Golovko, A., Adams, J., Guo, H. et al. Consistent Production of Mice with Conditional Knockout Alleles by CRISPR/Cas9-Mediated Genome Editing Using Two Guides/Two Oligos Approach. Cytol. Genet. 54, 63–70 (2020). https://doi.org/10.3103/S0095452720010065
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3103/S0095452720010065