
Abstract
The effect of the nature of acids on the kinetic regularities of the dissolution of iron (hydr) oxide phases in acidic solutions is studied. The nature of the potential-determining reaction in the magnetite electrode/orthophosphoric acid solutions system is revealed. It is shown based on the experimental data that the dissolution of iron oxide phases can be stimulated varying the iron oxide/electrolyte solution phase-boundary potential.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The determination of the composition and stability constants of complexes of metal orthophosphates generally and iron orthophosphates specifically are difficult tasks as shown by the limited and contradictory published data [1–4].
The review [4] summarizes the data on the complexation of orthophosphate anions with iron(II) and iron(III) cations in aqueous solutions, as well as analyzes protolytic reactions in the orthophosphoric acid–water system. At [H+] > 10–6 mol L–1 [4–6], aqueous solutions of H3PO4 mainly contain dihydrophosphate ions and orthophosphoric acid molecules. In concentrated solutions of H3PO4, the portion of dimers and their dissociation products is high [5, 7]. The constants for all the considered molecules of orthophosphoric acid are given in Table 1 [4, 5]. They are in accordance with the earlier obtained values [1–3, 8–11]; however, in some cases a slight deviation from the data of [4] are observed [4].
The experimental data on the complexation of Fe(II) and Fe(III) cations with orthophosphate anions were obtained at 25°C and an ionic strength of 3 mol L–1 (NaClO4); the constants were calculated also for infinitely diluted solutions [4, 12–14]. Polynuclear complexes were identified for ferric iron [4, 6, 13, 14]. It should be noted that their presence at pH < 0.5–1.0 is insignificant with the concentration of orthophosphate ions being predominant [4, 6]. The kinds of Fe(II) and Fe(III) orthophosphate complexes and their stability constants are given in Table 2 [4].
Some Fe(II) orthophosphate complexes (1 : 1) described in [4] have been obtained earlier [1–3]. The studies of [4, 15] give the values of constants for \({\text{Fe}}{{{\text{H}}}_{2}}{\text{PO}}_{4}^{ + }\) and FeHPO4 complexes (at infinite dilution). The differences in the values of constants are likely due to a low stability of these complexes [4].
The data on the complexation in the Fe(III)–orthophosphoric acid system are given in [4, 13, 14, 16–22]. The differences in the constant values in different authors are due to different experimental conditions, as well as due to the fact that the constants for mononuclear complexes were determined by the data obtained for solutions containing polynuclear forms of complexes [4, 13, 14]. Also, it should be noted that the reaction of Fe(II) and Fe(III) ions with orthophosphate ions is likely to occur through the elimination of the dihydrophosphate ion from the dimeric forms of orthophosphoric acid followed by complexation with monomeric ions [23, 24]. The data of these studies show that the nature of the complexes is significantly influenced by the concentration of Fe(III) ions (mono- or polyforms of complexes), H+, and phosphate ions (different degree of protonation of the ligand and different coordination numbers of complexing agent).
Thus, Fe(II) and Fe(III) phosphate complexes in aqueous solutions are products of the sequential replacement of water molecules with hydrophosphate and dihydrophosphate ions in iron aquacomplexes. The increase in the concentration of orthophosphoric acid in a solution and, consequently, pH leads to an increase in the coordination number of the complexing agent and degree of protonation of the ligand [4, 6, 24–27]. The coordination number of the complexing agent does not exceed three [4, 6, 14, 15, 25]. No hydrolysis and polymerization of Fe(II) and Fe(III) ions were observed under the selected experimental conditions (the concentration of H3PO4 was >1 mol L–1) [6, 23, 28].
The stability constants for the Fe(II) and Fe(III) complexes given in Table 2 are described by the interaction of the metal ion with dihydrophosphate ions and orthophosphoric acid, respectively [4].
The rate of dissolution of the oxide phases is governed by the nature and potential jump at the oxide/electrolyte solution interface. The potential-determining reactions in the multistep dissolution of oxides are quasi-equilibrium as it follows from the determination of the exchange current (10–1–10–3 A/cm2) of the potential-determining reactions (by Fe(III) and Fe(II) ions) depending on the anionic composition of the electrolyte, which considerably exceeds the dissolution current of the iron oxides (10–7–10–8 A cm–2) [29–31].
The study of the magnetite electrode potential as a function of some factors [29, 31] showed that, in strongly acidic media (pH < 1) in the presence of Fe(III) and Fe(II) ions, the potential-determining reaction is a process described by the equation that taking into account the presence of complexing agents (A) in solutions will be as follows:
The dissolution current of magnetite as a function of potential was studied in [29, 32]. The increase in the dissolution current was found for iron oxides upon the cathode polarization of the magnetite electrode in acidic media, including orthophosphoric acid.
The aim of this study is to establish and explain the relationship between the rate of dissolution of iron oxide phases in orthophosphoric acid (compared to other acidic solutions) and the complexation of Fe(II) and Fe(III) ions with orthophosphoric acid and its anions.
EXPERIMENTAL
In the kinetic study of dissolution, the main test objects were the iron oxide phases: α-Fe2O3 (hematite) and Fe3O4 (magnetite and iron hydroxide α-Fe2O3⋅xH2O). The samples were identified by powder X-ray diffraction. All reagents used were of the analytical grade. Iron (hydr)oxide (0.100 g) was placed in a temperature-controlled cell (TS-16 thermostat) containing a solution of electrolyte(s) (0.500 ± 0.005 L). The empirical kinetic dissolution curves were obtained for temperatures of 293 and 313 K. To eliminate diffusion hindrances, (hydr)oxide was dissolved in the mode of mechanical stirring of the water-dispersed mixture at 600–700 rpm [29–3131]. A portion of the solution was sampled from the cell at intervals and the total concentration of the iron(II) ions (the iron(III) ions were preliminarily reduced with hydroxylamine) was determined by photocalorimetry on a KFK-3-01 photometer using o-phenanthroline [33]. The primary analysis of the kinetic data was performed in the following coordinates: portion of dissolved oxygen (α) – time (t). The portion of dissolved oxygen (α was calculated by the equation α = A/A∞, where A and A∞ are the absorbances of the filtrate’s solution at time t and upon the complete dissolution of the iron oxide phase, respectively [29–31, 34].
To study the electrochemical behavior of magnetite, an electrode was made of this material according to the procedure of [35]. The reference electrode was a saturated silver chloride electrode (the potential vs. the normal hydrogen electrode at 20°C was 0.201 ± 0.003 V). The potential was measured using a pH meter (pH-150MP.2 millivoltmeter) in the standard electrochemical cell at 22 ± 0.5°C in solutions of electrolytes.
The complexation of high-concentration solutions of orthophosphoric acid with iron(III) ions (0.16 g L–1) was studied by UV-vis spectrophotometry. The data were obtained using a SHIMADZU UV-1700 PharmaSpec UV-VIS spectrophotometer. Spectra were recorded relative to water and solutions of orthophosphoric acid at a certain concentration using quartz cuvettes (1 mm). For the UV-vis spectrophotometric study, solutions of orthophosphoric acid (1.23, 2.46, 3.69, 4.92, and 6.15 M) containing a certain amount of iron(III) ions were used. The solutions were prepared dissolving the iron oxide phase, hematite, in orthophosphoric acid.
RESULTS AND DISCUSSION
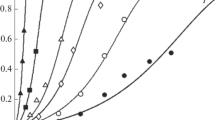
The data obtained in the empirical studies of the kinetic dependences of dissolution of the iron oxide phases, i.e., Fe3O4, α-Fe2O3, and hydrated α-Fe2O3, in different media as a function of time are given in Figs. 1 and 2.

Portion of dissolved oxide (α) as a function of time (t) for Fe3O4 (a) in H2SO4 (3) (2.50 М, f = 1/2), HCl (2) (5.00 M, f = 1/1), and H3PO4 (1) (1.67 M, f = 1/3) acids at 293 K, for α-Fe2O3 (b) in HClO4 (4) (1.00 М, f = 1/1), H2SO4 (3) (0.50 M, f = 1/2), HCl (2) (1.00 M, f = 1/1), and H3PO4 (1) (0.33 М, f = 1/3) acids at 313 K and for hydrated α-Fe2O3 (c) at 313 K in HClO4 (5) (1.00 M, f = 1/1), HNO3 (4) (1.00 M, f = 1/1), H2SO4 (3) (0.50 M, f = 1/2), HCl (2) (1.00 M, f = 1/1), H3PO4 (1) (0.33 М, f = 1/3). The points are experimental data and the lines are graphic presentation of the heterogeneous kinetics equation –ln(1 – α) = Ash(Wi t) = Ash(τ).

Portion of dissolved magnetite (α) as a function of time (t) upon dissolution in acids and their mixtures (T = 313 K, the concentration of all acids was 6.50 N): (a) H3PO4 (1), CH3COOH + H3PO4 (1 : 1) (2), H2SO4 + H3PO4 (1 : 1) (3), H2SO4 (4), CH3COOH (5); (b) H3PO4 (1), H2SO4 + H3PO4 (1 : 9) (2), H2SO4 + H3PO4 (1 : 4) (3), H2SO4 + H3PO4 (1 : 1) (4), H2SO4 + H3PO4 (4 : 1) (5), H2SO4 (6). The points denote experimental data and the lines are graphic presentation of the heterogeneous kinetics equation –ln(1 – α) = Ash(Wi t) = Ash(τ).
To reveal how the acid mixtures influence the rate of dissolution of magnetite, the effect of H3PO4 additions to CH3COOH and, in more detail, to H2SO4 was studied.
It is seen from the given experimental data that the rate of dissolution of the iron (hydr) oxide phases is higher in solutions containing orthophosphoric acid; and the rate increases with an increase in the acid concentration [29, 31, 34].
The experimental data for the dependence of the magnetite electrode’s potential on the concentration of ions in solutions of orthophosphoric acid and its mixture with acetic and sulfuric acids (Fig. 3) confirm that there is a correlation between the kinetic dependence and potentiometric data. The obtained data do not contradict those given in [29, 36–39].

Potential of magnetite electrode (E, V) as a function of the logarithmic concentration of ions (log C; C, M): (a) iron(III), (b) H+, (c) orthophosphoric acid, and (d) iron(II).
The analysis of the data shows that, upon an increase in the concentration of the orthophosphoric acid (the portion of the orthophosphoric acid in the acid mixture), the potential of the magnetite electrode shifts to negative values due to the complexation of the orthophosphoric acid and its ions with Fe(III) ions to a higher degree compared to the Fe(II) ions (Table 3). This agrees with the kinetic data on an increase in the rate of dissolution of oxide phases and the effect of potential on their dissolution rate [29–31, 34].
The potential-determining reaction in the magnetite electrode–orthophosphoric acid solution system is governed by four parameters: the concentrations of the H+, iron(II), and iron(III) ions, as well as the concentration of the orthophosphoric acid. The dependences of the magnetite electrode’s potential on the logarithmic concentration of the above-mentioned ions are shown in Fig. 3.
As the given data show, the potential increases with an increase in the concentration of iron(III) and a decrease in pH, and the potential decreases with an increase in the concentrations of the orthophosphoric acid and iron(II) ions.
To clarify the nature of the potential-determining reaction in the magnetite–orthophosphoric acid solution system, it is necessary to know how the distributions of portions of Fe(III) and Fe(II) ions, as well as their phosphate complexes in solutions of orthophosphoric acid, depend on the acid concentration. Taking into account that upon the dissolution of the iron oxide phases the concentration of the ligand (orthophosphoric acid and its anions) is multiple times higher than that of the complexing agent (Fe(III) and Fe(II)), we can assume that [Fe(H2PO4)3]0 and [Fe(H2PO4)2]0 predominate in the solution of orthophosphoric acid upon dissolution of the oxide phase [4, 12–14]. The existence of only one complex in the studied solutions of orthophosphoric acid is confirmed by the data of the UV-vis spectrophotometric analysis (Fig. 4).

(I) Electronic absorption spectra for solutions of orthophosphoric acid (the acid concentration was 1.23 (1), 2.46 (2), 3.69 (3), 4.92 (4), and 6.15 M (5)) and solutions of orthophosphoric acid (6.15 (6), 3.96 (7), 4.92 (8), 2.46 (9), and 1.23 M (10)) containing iron(III) ions (0.16 g L–1) recorded relative to water. (II) Electronic absorption spectra for solutions of orthophosphoric acid (the acid concentration was 3.69 (1), 6.15 (2), 4.92 (3), 1.23 (4), and 2.46 M (5)) containing iron(III) ions (0.16 g L–1) recorded relative solutions of orthophosphoric acid with the same concentration.
CONCLUSIONS
Based on the reported and considered data, we can assume that the potential-determining reaction in the magnetite–orthophosphoric acid solution system is as follows:
REFERENCES
Sillén, L.G., Martell, A.E., and Bjerrum, A., Stability Constants of Metal-Ion Complexes, London; R. Soc. Chem., 1964.
Stability Constants of Metal-Ion Complexes, Part A: Inorganic Ligands, Högfeldt, E., Ed., Oxford; Pergamon, 1982.
The IUPAC Stability Constants Data Base, Pettit, G., Ed., Otley, UK: Academic Software, 1995.
Ciavatta, L., Recent Res. Dev. Inorg. Organomet. Chem., 2001, vol. 1, p. 83.
Ciavatta, L., Iuliano, M., Porto, R., and Vasca, E., Polyhedron, 1991, vol. 10, no. 22, p. 2587.
Al-Sogair, F., Marafie, H.M., Shuaib, N.M., Youngo, H.B., and El-Ezaby, M.S., J. Coord. Chem., 2002, vol. 55, no. 9, p. 1097.
Elmore, K.L., Hatfield, J.D., Dunn, R.L., and Jone, A.D., J. Phys. Chem., 1965, vol. 69, no. 10, p. 3520.
Ciavatta, L. and Iuliano, M., Ann. Chim. (Rome, Italy), 1996, vol. 86, p. 1.
Pettersson, L., Acta Chem. Scand., 1959, vol. 197, no. 1, p. 25.
Baldwin, W.G. and Sillen, L.G., Ark. Kemi, 1969, vol. 31, p. 391.
Havel, J. and Hogfeldt, E., Chem. Scr., 1974, vol. 5, p. 164.
Ciavatta, L., Iuliano, M., and Porto, R., Ann. Chim. (Rome, Italy), 1992, vol. 82, p. 121.
Ciavatta, L. and Porto, R., Ann. Chim. (Rome, Italy), 1992, vol. 82, p. 447.
Ciavatta, L., Ann. Chim. (Rome, Italy), 1995, vol. 85, p. 235.
Nriagu, J.O., Geochim. Cosmochim. Acta, 1972, vol. 36, p. 459.
Sidorenko, V.I., Zh. Neorg. Khim., 1973, vol. 28, no. 5, p. 1270.
Filatova, L.N. and Galochkina, G.V., Zh. Neorg. Khim., 1974, vol. 19, no. 2, p. 3064.
Filatova, L.N., Zh. Neorg. Khim., 1974, vol. 19, no. 12, p. 3335.
Ramamoorthy, S. and Manning, P.G., Inorg. Nucl. Chem. Lett., 1974, vol. 10, no. 1, p. 109.
Filatova, L.H., Shelyapina, M.A., Blachinda, A.C., and Makarov, E.F., Zh. Neorg. Khim., 1976, vol. 21, no. 10, p. 2715.
Wilhelmy, R., Patel, R.C., and Matijevic, E., Inorg. Chem., 1985, vol. 24, p. 3290.
Khoe, G.H. and Robins, R.G., J. Chem. Soc., Dalton Trans., 1988, no. Iss. 8, p. 2015.
Filatova, L.N., Zh. Fiz. Khim., 1980, vol. 54, no. 1, p. 179.
Filatova, L.N., Tr. IREA, 1974, no. 36, p. 164.
Filatova, L.N., Vendilo, A.G., and Sandu, R.A., Zh. Neorg. Khim., 2012, vol. 57, no. 9, p. 1355.
Prodan, I.E., Eshchenko, L.S., and Pechkovskii, V.V., Zh. Neorg. Khim., 1990, vol. 35, no. 4, p. 843.
Prodan, I.E., Eshchenko, L.S., and Pechkovskii, V.V., Zh. Neorg. Khim., 1989, vol. 34, no. 7, p. 1860.
Prodan, I.E., Eshchenko, L.S., and Pechkovskii, V.V., Zh. Neorg. Khim., 1989, vol. 34, no. 8, p. 2012.
Gorichev, I.G., Kutepov, A.M., Gorichev, A.I., Izotov, A.D., and Zaitsev, B.E., Kinetika i mekhanizm rastvoreniya oksidov i gidroksidov zheleza v kislykh sredakh (Kinetics and Mechanism of Dissolution of Iron Oxides and Hydroxides in Acidic Media), Moscow: Ross. Univ. Druzhby Narodov, 1999.
Gorichev, I.G. and Kipriyanov, N.A., Russ. Chem. Rev., 1984, vol. 53, no. 11, p. 1039.
Kuzin, A.V., Gorichev, I.G., and Lainer, Yu.A., Russ. Metall. (Engl. Transl.), 2013, no. 5, p. 652.
Kuzin, A.V., Gorichev, I.G., Batrakov, V.V., and Lainer, Yu.A., Russ. Metall. (Engl. Transl.), 2014, no. 1, p. 33.
Marczenko, Z., Kolorymetryczne Oznaczanie Pierwiastkow (Colorimetric Determination of Elements), Warszawa: Wysdawnictwa Naukowo-Techniczne, 1968.
Kuzin, A.V., Gorichev, E.A., and Eliseeva, L.E., Slynko, Vestn. Mosk. Gos. Tekh. Univ. im. N. E. Baumana, Ser. Estestv. Nauki, 2019, no. 2, p. 104.
Gorichev, I.G. and Mikhal’chenko, I.S., Zashch. Met., 1989, vol. 25, no. 4, p. 577.
Zakharov, V.A., Songina, O.A., and Berkutova, G.B, Zh. Anal. Khim., 1976, vol. 31, no. 11, p. 2212.
Avdeev, Ya.G. and Andreeva, T.E., Usp. Khim. Khim. Tekhnol., 2018, vol. 32, no. 13, p. 63.
Avdeev, Ya.G., Andreeva, T.E., Panova, A.V., and Kuznetsov, Yu.I., Int. J. Corros. Scale Inhib., 2019, vol. 8, no. 1, p. 139.
Avdeev, Ya.G., Panova, A.V., Andreeva, T.E., and Kuznetsov, Yu.I., Korroz.: Mater., Zashch., 2019, no. 11, p. 32.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
There are no supplementary materials.
This article does not contain studies with the use of humans or animals as research objects.
Additional information
Translated by K. Utegenov
About this article

Cite this article
Kuzin, A.V., Gorichev, I.G., Shelontsev, V.A. et al. The Role of a Complex Formation in the Dissolution of Iron Oxides in Orthophosphoric Acid. Moscow Univ. Chem. Bull. 76, 398–404 (2021). https://doi.org/10.3103/S0027131421060055
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3103/S0027131421060055