
Abstract
Laccases (EC 1.10.3.2) are multicopper polyphenol oxidases found in various organisms, in particular, in fungi. Fungal laccases are encoded by multigene families, which can contain up to 17 genes. However, not all isozymes can be obtained from native producers. Previous studies have shown that the filamentous fungus Penicillium canescens is a promising object for the heterologous expression of various laccase isozymes of Trametes hirsuta 072. In this work, the cultivation conditions of P. canescens strains, recombinant producers of T. hirsuta heterologous minor laccase, are optimized. The optimization of the rLacD and rLacF purification method increased their yield and specific activity of rLacD. In addition, the melting points of minor laccase isoenzymes are measured and the glycosylation of isoenzymes is studied.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The problem of recycling of lignocellulosic biomass (wood, straw, stalks grain, etc.) is relevant today. The recycling process largely depends on the modification (destruction) of lignin, which is part of lignocellulose—a complex biopolymer with an irregular structure, which is a mixture of aromatic mono- and oligomers. In natural ecosystems, fungi are the main modifyers of lignocellulosic biomass, with more than 90% of the lignocellulose being degraded by basidiomycetes in basidiomycetes [1]. It is well known that their ability to decompose lignocellulose is provided by the action of a unique complex of extracellular enzymes, in which laccases play a key role (EC 1.10.3.2) [2]. Laccases are fairly stable enzymes, with broad substrate specificity for various phenolic compounds [4, 5] and the ability to reduce oxygen without the formation of peroxide. In addition, they can modify various xenobiotics (for example, herbicides and dyes), which are also in demand in various branches of biotechnology [4–6]. Noted that the demand not only for laccases with a broad substrate specificity but also for more specific enzymes capable of selectively modifying certain components of complex mixtures has been growing recently [7]. Currently, the biotechnological potential of laccases has not been fully disclosed. Primarily, this is due to the limited knowledge about their physicochemical and catalytic properties, as well as the lack of highly active producers. The accumulated data on the genome-wide sequencing of higher fungi showed that laccases are encoded by multigene families, which can reach 17 members [8]. At the same time, the core production of proteins by the native producer is usually limited to a small spectrum of laccase isoenzymes.
White rot basidiomycetes of the genus Trametes is a promising object for study. As a rule, representatives of this genus produce one constitutive (major) laccase isoenzyme under almost all cultivation conditions [9–11]. The remaining isoenzymes are inducible (minor) forms that differ from the major ones by their biochemical properties [11–15]. The production of minor isoenzymes using native producers is usually difficult due to the difficulty of selecting the cultivation conditions and inductors necessary for their biosynthesis. For this reason, in the majority of the fungi of this genus, only the major laccase isoenzymes have been sufficiently studied [16, 17]. Thus, the search for new laccase isoenzymes of basidiomycetes and a comparative study of their properties is an urgent task in modern biochemistry, not only from a practical point of view but also from a fundamental one.
Basidiomycete Trametes hirsuta 072 is an effective lignin destructor. Its multigenic laccase family is represented by seven genes (lacA, lacB, lacC, lacD, lacE, lacF, and lacG) [18].
In previous studies, in addition to the native major isoenzyme LacA, we were also able to obtain recombinant minor rLacC, rLacD, and rLacF isoenzymes [19–21] in Penicillium canescens ascomycete and partially characterize them. The purpose of this study is to optimize the cultivation conditions of recombinant strains-producers of minor laccases, optimize the purification conditions of the target enzymes, and study their properties.
MATERIALS AND METHODS
Strains and Media Used
We used P. canescens Cc1(25)25, P. canescens Dc2(6)23, and P. canescens Fc3(5)27 strains—producers of recombinant isoenzymes rLacC, rLacD, and rLacF T. hirsuta 072, respectively, which were previously obtained by us [17, 22].
The strains were grown as described in [23] at different pH values (4.6; 5.6; 7.0) and different concentrations of copper sulfate (0.001–2 mM). To study the kinetics of their enzymatic activity, the strains were grown for 8 days under the chosen conditions and samples were taken every 24 h.
Determination of Enzyme Activity
The activity of isoenzymes was determined spectrophotometrically using a PerkinElmer Lambda 35 spectrophotometer (United States), as described in [22]. A solution of 2,2-azino-bis(3-ethylbenzthiazolin-6-sulfonic acid) diammonium salt (ABTS, λ = 436 nm, ε = 29 500 M–1 cm–1) was used as the chromogenic substrate. An increase in the optical density in 1 mL of the reaction mixture per minute was taken as the conventional unit of activity. The specific activity was calculated for 1 mg of protein.
Determination of Protein Concentration
Samples of the culture broth (CB) were centrifuged at 14 000 rpm for 5 min. The protein concentration in the supernatant was determined spectrophotometrically using a commercial BCA Protein Assay Kit (Pierce, United States), according to the manufacturer’s method.
Purification of Isoenzymes
The initial scheme (1) of purifying recombinant isoenzymes is described in [17, 22]. The optimized purification scheme (2) included the following stages: the filtrate of CB was concentrated using the Millipore Pellicon 2 tangential flow ultrafiltration system (Merck Millipore, United States) using a cellulose membrane. The ultrafiltrate was then dialyzed against a buffer solution (5 mM Tris Acetate buffer (pH 8.0) was used for rLacD, 5 mM Bis-Tris (pH 6.0) was used for rLacF), applied onto a Source 15Q column (GE Healthcare, Uppsala, Sweden), and equilibrated with an appropriate buffer solution. Elution was performed in a sodium chloride salt gradient (5 mM Tris-acetate buffer (pH 8.0) to a 5 mM Tris-acetate buffer + 400 mM NaCl (pH 8.0) was used for rLacD; 5 mM Bis-Tris (pH 6.0) to a 5 mM Bis-Tris + 400 mM NaCl (pH 6.0) was used for rLacF). The most active fractions were combined and concentrated. In the next step, the enzymes were dialyzed against a sodium acetate buffer solution (30 mM, pH 5.0) were applied onto a Source 15ISO column (GE Healthcare, Uppsala, Sweden), equilibrated with a 1.7 M (NH4)2SO4 + 30 mM Na-acetate buffer solution (30 mM, pH 5.0), and eluted with a 30 mM sodium acetate buffer (pH 5.0). The active fractions were combined, concentrated, and used for further study.
Analysis of Protein Fractions
The molecular weight (MW) of the isoenzymes was determined using SDS electrophoresis in a 12% polyacrylamide gel, as described in [22]. To determine the isoelectric point (IEP, pI) of the proteins, isoelectric focusing was performed in a polyacrylamide gel using pH 3.0–10.0 ampholytes (Serva Electrophoresi, Germany) on a 111 Mini IEF Cell device (Bio-Rad), United States). The pH gradient was measured using the calibration standards (GE Healthcare, United Kingdom). Coomassie Brilliant Blue R-250 (AppliChem, Germany) was used for protein staining. The calculated molecular weights and pI of the isoenzymes were determined by known amino acid sequences using the ExPASy service (https://www.expasy.org/) [24].
Analysis of Glycosylation Sites
The location of the glycosylation sites of laccase isoenzymes was calculated in accordance with the known amino acid sequences using the NetNGlyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) service [25]. For the experimental confirmation of their presence, a fragment of a protein band corresponding to the target isoenzyme was cut out of the gel obtained after SDS electrophoresis, and then, using the standard protocol [26], protein hydrolysis was performed. The resulting peptides were extracted from the gel with a 20% acetonitrile solution containing 0.1% trifluoroacetic acid, and were determined by MALDI-TOF/ TOF MS spectrometry using an Ultraflextreme mass spectrometer (Bruker, Germany). Protein identification was performed using the Mascot program (www.matrixscience.com). For the processing of mass spectra, the software package FlexAnalysis 3.3 (Bruker Daltonics, Germany) was used.
Study of the Thermal Stability of Isoenzymes Using Differential Scanning Calorimetry (DSC)
The melting point (Tm) of the laccase was determined using an N-DSC III nanodifferential scanning calorimeter (TA Instruments, United States) at a heating rate of 1°C/min, at 30–95°C and an overpressure of 3.0 atm. The protein concentration was 1.2 mg/mL. Basic adjustments were made and smoothed by subtracting the buffer thermogram. The data was analyzed on a Launch NanoAnalyze Software instrument (TA Instruments, United States). The calorimetric heat (ΔHcal) was calculated from the size of the area under the curve of the dependence of the excess heat capacity of protein on temperature, and the melting point (Tm) was determined as the maximum temperature of the melting curve’s peak.
The relative error in the calculation of ΔHcal was 5–8%. The experimental measurement error Tm did not exceed ±0.2°.
RESULTS AND DISCUSSION
Optimization of Cultivation Conditions of Producers
One of the simplest methods of stimulating the synthesis of laccase and increasing its activity is the addition of copper ions to the medium, usually in the form of CuSO4 [27, 28]. However, in an excessively high concentration, copper ions are extremely toxic to cells, primarily because of their ability to participate in the formation of hydroxyl radicals, leading to the destruction of cellular structures [29, 30]. Therefore, in the present work, the optimal concentration of CuSO4 in the medium was determined in order to avoid inhibiting the growth of the fungus and simultaneously stimulate the production of active target enzymes. Copper sulfate was added to the growth medium of laccase producers in a concentration ranging from 0.001 to 2 mM. It was shown that the maximum activity of all target isoenzymes is achieved using 0.5 mM CuSO4 and is 190, 104, and 150 UE/mL for rLacC, rLacD, and rLacF, respectively. With other concentrations of CuSO4 (in the studied range), the activity of isoenzymes decreased without changing the growth rate of the culture, except for a concentration of 2 mM (in this case growth was inhibited).
In addition, the optimum initial pH value of the medium was selected. Three initial pH values were used (4.6, 5.6, 7.0). The highest activity of the target proteins was achieved at pH 4.6 for all isoenzymes. This result is consistent with the published data, since previous studies have shown that the optimum pH values for fungal laccases are in the pH range from 4 to 6 [6]. It should be noted that in the process of cultivation (at the initial pH of 4.6) there were differences in the rate of change in the pH of the growth medium. However, after 6 days, the pH of the medium was about 7.0 for all the strains studied. The same trend continued for the recipient strain. It is known that alkalization of the medium during cultivation is characteristic of filamentous fungi of the Penicillium genus and is associated with the deamination of amino acids and the subsequent formation of ammonia [31].
The optimal duration of the cultivation of strains for the isolation of target isoenzymes in the chosen conditions was determined within 8 days (Fig. 1). Figure 1 shows that for all strains a significant increase in the activity of the target enzymes occurred after 4 days of cultivation up to 8 days. However, after 6 days of cultivation, all strains showed intense autolysis of cells; therefore, a duration of 6 days was chosen as the optimal one. On the 6th day, the activity of rLacC and rLacF isoenzymes in CB reached values of 150 to 190 UE/mL, and the activity of rLacD was one-and-a half lower.

Kinetics of laccase activity change depending on duration of cultivation of strains: 1, rLacC; 2, rLacD; 3, rLacF (average values are given for three technical replicates, error does not exceed 3%).
Purification and Properties of Minor T. hirsuta 072 Laccase Isoenzymes
Previously, we developed a purification scheme for isolating the major native LacA from the CB of T. hirsuta 072 [19].
This scheme (with some modifications) was also used for the purification of recombinant minor rLacC, rLacD, and rLacF [17, 22] (Table 1, Scheme 1). However, it was shown that the specific activity of the rLacD and rLacF isoenzymes (40 and 41 UE/mg, respectively) isolated according to this scheme was significantly lower compared to rLacC (260.3 UE/mg). It can be seen that the main limiting stage in Scheme 1 is the precipitation of proteins with ammonium sulfate. Therefore, in the present work, at the first stage of rLacD and rLacF isolation, CB ultrafiltration in the tangential flow on a cellulose membrane was used to concentrate the proteins. In addition, ion exchange chromatography was carried out on a Source 15Q exchanger, and hydrophobic chromatography on a Source 15ISO exchanger was used instead of gel permeation chromatography (Table 1, Scheme 2).
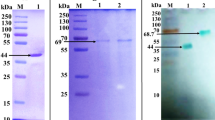
Optimization of the purification scheme reduced the number of stages of isolation of isoenzymes rLacD and rLacF, while their output increased to 13 and 12%, respectively, of the total activity in the original CB. It was also possible to increase the specific activity of the obtained isoenzyme rLacD to 63 UE/mg. The specific activity of rLacF did not change. Mass spectrometric identification of the proteins obtained confirmed that they are the rLacC, rLacD, and rLacF isoenzymes. All the obtained isoenzymes in the solution had a blue color of varying intensity, typical of the classic blue laccases [32]. The molecular weight and isoelectric points of the isoenzymes differed from those predicted (Fig. 2, Table 2).

Electrophoretic analysis of laccase preparations: (a) SDS-electrophoresis under denaturing conditions (1, 6, Molecular weight marker; 2, LacA; 3, rLacC; 4, rLacF; 5, rLacD); (b) isoelectric focusing of laccase isoenzymes (1, LacA; 2, rLacC; 3, rLacF; 4, rLacD; 5, marker).
As can be seen from the presented data, the LacA and rLacC isoenzymes have similar molecular properties (MW and pI), which made it possible to successfully apply the scheme developed for purifying LacA to the recombinant isoenzyme rLacC. However, the values of MW and pI of the other two minor isoenzymes (rLacD and rLacF) are significantly different. In addition, when growing the producer of rLacF, the obtained CB turned out to be more pigmented compared to the two other producers (rLacD and rLacC), which also made it difficult to purify this isoenzyme according to Scheme 1.
As can be seen from Fig. 2, the MW values of the obtained recombinant enzymes differed from the calculated values (Table 2), while the MW values of rLacD and rLacF were significantly higher than for rLacC. Based on the data obtained, the degree of glycosylation of isoenzymes was 17, 20, and 23% for rLacC, rLacF, and rLacD, respectively.
Thus, it was found that the obtained recombinant isoenzymes rLacD and rLacF have a greater degree of glycosylation than rLacC and the previously studied LacA [11], which may be due to the presence of additional glycosylation sites on the surface of the protein globules. Analysis of the amino acid sequences of the rLacD, rLacC, and rLacF isoenzymes showed that the calculated number and location of the N-glycosylation sites of T. hirsuta 072 laccases are noticeably different, and for LacF the number of calculation sites is half that for the other two minor isoenzymes.
Figure 3 shows a fragment of the alignment of the amino acid sequences of the isoenzymes of T. hirsuta 072 laccases in the region of amino acid residues where the N-glycosylation sites are located. As can be seen from Fig. 3, for the T. hirsuta 072 laccase isoenzymes studied in this paper, two glycosylation sites are conservative (Asn75 and Asn457). At the same time, the presence of only Asn75 was experimentally confirmed for all the studied laccase isoenzymes. Interestingly, for rLacC and rLacD, we experimentally confirmed the presence of such unique sites as Asn207 and Asn292, respectively (Fig. 3), which were not found in other fungal laccases described in the literature. It should be noted that in the structure of LacA isolated from a natural producer (PDB: 3FPX) [19], four of the eight possible glycosylation sites were found (Table 2), and in the structure of recombinant rLacA (PDB: 5LDU), obtained earlier in P. canescens [23], the presence of an additional Asn398 N-glycosylation site was confirmed, in addition to the sites confirmed for LacA. The presence of this glycosylation site was experimentally confirmed for rLacF (Fig. 3, Asn402).

Sites of N-glycosylation of laccase isoenzymes LacA (GenBank: KP027478.1), LacC (GenBank: KP027479.1), LacD (GenBank: KP027480.1), LacF (GenBank: KP027482.1) of T. hirsuta 072: calculated glycosylation sites are underlined; asterisk (*, **) indicates conservative sites; gray, confirmed sites; bold, additional site, presence of which is shown for recombinant laccase A (native one is absent); frame indicates unique glycosylation sites, presence of which has been confirmed experimentally.
In [33], 3D structures of various laccase isoenzymes of the Pycnoporus sanguineus fungus also revealed differences in the location of N-glycosylation sites.
It is known that glycosylation of proteins affects their thermostability. In this respect, the thermal stability of minor laccase isoenzymes was studied in this work (Table 3). The melting curves of recombinant enzymes have two maxima (in contrast to the native laccase LacA), and the values of Tm of the first and second peaks are very close in all the studied enzymes, while the values of the melting heat (ΔHcal) vary greatly (Table 3). Note that the laccase rLacC, which is least stable at 60°C, has the highest ΔHcal value, but Tm of the first melting peak is 60°C, while for the remaining recombinant laccases this indicator shifts by 1–2°C to higher temperatures. In native LacA laccase, the first melting peak is absent, and the value of the melting heat is the lowest of all the isoenzymes studied.
Thus, it can be assumed that the differences in the physicochemical and catalytic properties of laccase isoenzymes, shown here and earlier [17, 22], are probably due to the glycosylation pattern of the isoenzymes.
However, we cannot unequivocally state that the characteristics obtained for recombinant minor isoenzymes are similar to the characteristics of isoenzymes from a natural producer. Therefore, the search for ways to obtain native minor laccase isoenzymes of T. hirsuta 072 remains an urgent task.
REFERENCES
Baldrian, P., FEMS Microbiol. Rev., 2006, vol. 30, no. 2, p. 215.
Have, T.R. and Teunissen, P.J.M., Chem. Rev., 2001, vol. 101, no. 11, p. 3397.
Riva, S., Trends Biotechnol., 2006, vol. 24, no. 5, p. 219.
Khan, R., Bhawana, P., and Fulekar, M.H., Rev. Environ. Sci. Biotechnol., 2013, vol. 12, no. 1, p. 75.
Chandra, R. and Chowdhary, P., Environ. Sci. Process. Impacts, 2015, vol. 17, no. 2, p. 326.
Viswanath, B., Rajesh, B., Janardhan, A., et al., Enzyme Res., 2014, vol. 2014, 163242.
Minussi, R.C., Pastore, G.M., and Duran, N., Trends Food Sci. Technol., 2002, vol. 13, nos. 6–7, p. 205.
Kilaru, S., Hoegger, P.J., and Kües, U., Curr. Genet., 2006, vol. 50, no. 1, p. 45.
Jia, L.D., Zhang, Y.W., Zhang, R.H., et al., J. Basic Microbiol., 2005, vol. 45, no. 3, p. 190.
Lorenzo, M., Moldes, D., and Sanromán, M.Á., Chemosphere, 2006, vol. 63, no. 6, p. 912.
Vasina, D.V., Mustafaev, O.N., Moiseenko, K.V., et al., Biochimie, 2015, vol. 116, p. 154.
Yaver, D.S., Xu, F., Golightly, E.J., et al., Appl. Environ. Microbiol., 1996, vol. 62, no. 3, p. 834.
Temp, U., Zierold, U., and Eggert, C., Gene, 1999, vol. 236, no. 1, p. 169.
Xiao, Y.Z., Chen, Q., Hang, J., et al., Mycologia, 2004, vol. 96, no. 1, p. 26.
Koschorreck, K., Richter, S.M., Swierczek, A., et al., Arch. Biochem. Biophys., 2008, vol. 474, no. 1, p. 213.
Yang, J., et al., Front. Microbiol., 2017, vol. 8.
Savinova, O.S., Moiseenko, K.V., Vavilova, E.A., et al., Front. Microbiol., 2019, vol. 10, no. 2. https://doi.org/10.3389/fmicb.2019.00152
Pavlov, A.R., Tyazhelova, T.A., Moiseenko, K.V., et al., Genome Announce., 2015, vol. 3, no. 6, e01287-15.
Polyakov, K.M., Fedorova, T.V., Stepanova, E.V., et al., Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, vol. 65, no. 6, p. 611.
Rebrikov, D.V., Stepanova, E.V., Koroleva, O.V., et al., Appl. Biochem. Microbiol., 2006, vol. 42, no. 6, p. 564.
Glazunova, O., Trushkin, N.A., Moiseenko, K.V., et al., Catalysts, 2018, vol. 8, no. 4, p. 152.
Savinova, O.S., Moiseenko, K.V., Vavilova, E.A., et al., Biochimie, 2017, vol. 142, p. 183.
Abyanova, A.R., Chulkin, A.M., Vavilova, E.A., et al., Appl. Biochem. Microbiol., 2010, vol. 46, no. 3, p. 313.
Wilkins, M.R., Gasteiger, E., Bairoch, A., et al., Methods Mol. Biol., 1999, vol. 112, p. 531.
Gupta, R. and Brunak, S., Biocomputing, 2002, vol. 322, p. 310.
Smith, B.J. and Tempst, P., in Methods in Molecular Bio-logy, vol. 64: Protein Sequencing Protocols, Smith, B.J., Ed., Totowa, NJ: Humana, 1996, p. 1.
Huber, M. and Lerch, K., FEBS Lett., 1987, vol. 219, no. 2, p. 335.
Palmieri, G., Giardina, P., Bianco, C., et al., Appl. Environ. Microbiol., 2000, vol. 66, no. 3, p. 920.
Arora, D.S. and Rampal, P., J. Basic Microbiol., 2002, vol. 42, no. 5, p. 295.
Galhaup, C. and Haltrich, D., Appl. Microbiol. Biotechnol., 2001, vol. 56, nos. 1–2, p. 225.
Paveia, M.H., Mycopathologia, 1975, vol. 55, p. 35.
Morozova, O.V., Shumakovich. G.P., Gorbacheva, M.A., et al., Biochemistry (Moscow), 2007, vol. 72, no. 10, p. 1136.
Orlikowska, M., de J. Rostro-Alanis, M., Bujacz, A., et al., Int. J. Biol. Macromol., 2018, vol. 107, p. 1629.
Glazunova, O.A., Shakhova, N.V., Psurtseva, N.V., et al., PLoS One, 2018, vol. 13, no. 6, e0197667.
ACKNOWLEDGMENTS
The authors thank S.Yu. Kleimenov, a member of the Laboratory for Protein Structural Biochemistry at the Bach Institute of Biochemistry, Research Center of Biotechnology, Russian Academy of Sciences, for his assistance in conducting experiments on differential scanning calorimetry.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they do not have a conflict of interest.
Additional information
Translated by P. Kuchina
About this article

Cite this article
Savinova, O.S., Zorov, I.N., Vasina, D.V. et al. The Minor Recombinant Laccase Isozymes of Trametes hirsuta 072: Preparation and Properties. Moscow Univ. Chem. Bull. 74, 173–179 (2019). https://doi.org/10.3103/S0027131419040072
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3103/S0027131419040072