
Abstract
Electrochemically reduced graphene oxide (ERGO) is a promising material for heavy metal ion detection due it its facile synthesis and electroactive defects. A simple ERGO sensor was fabricated directly on a glassy carbon electrode using cyclic voltammetry. Deposition cycles were optimized for sensitivity to the analytes, with 12 cycles found to be ideal. Differential pulse voltammetry was used to detect trace amounts of lead and cadmium, down to 5 nM for lead and 75 nM for cadmium (S/N > 3). The sensors were robust and their data reproducible, even in the presence of iron and zinc ions.
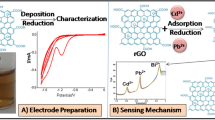
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Heavy metal ion (HMI) contamination is an urgent worldwide concern in terms of human and environmental safety. HMI such as mercury, lead, cadmium, and chromium are highly toxic and cause acute or chronic health problems in humans depending on exposure level,[[1]] and can stunt the growth of crops when present in soil, water, or air.[[2]] Other HMI, such as iron, zinc, and copper, are essential micronutrients in small amounts but can be toxic in large amounts; however, their relative harmlessness leads them to be ubiquitous in the environment. All three are commonly found in fertilizers, which when overused can lead to dangerous levels of accumulation in plants, water, and soil.[[3, 4]] Furthermore, the concentration of any HMI can spike dangerously due to human causes, such as improper disposal of industrial waste, leaching from infrastructure, or acid mine drainage;[[5]] thus, rapid and continuous detection of HMI is of utmost importance. This paper will focus on lead (Pb2+) and cadmium (Cd2+) due to their common industrial use and high toxicity.
Electrochemical techniques are a promising method for HMI detection in environmental samples. Although current analytical methods such as atomic absorption spectrometry (AAS) and inductively coupled plasma mass spectrometry (ICP-MS) can quantify trace amounts of HMI, they are expensive and unsuited to be adapted for portable and in situ applications.[[6]] Electrochemical techniques, meanwhile, are rapid, sensitive, and inexpensive. Essentially, when subjected to electric potential changes, redox active HMI will oxidize on the surface of the working electrode (WE). This oxidation results in a rapid increase of current, which is correlated to the concentration of HMI present in solution. These oxidation peaks occur at characteristic oxidation potentials, so multiple HMI signals can be detected and quantified in a single voltammetric scan.
However, the sensitivity and selectivity of the sensor depend greatly upon the qualities of the WE surface. Traditional electrode substrates such as the glassy carbon electrode (GCE) yield low signals with poor resolution unless they are modified with some other nanomaterial.[[7]] One promising nanomaterial, reduced graphene oxide (RGO), is a stable form of graphene that contains irremovable oxygenous defects in the carbon lattice, such as hydroxyl and epoxy groups.[[8, 9]] These defects, while inhibiting the material’s conductive properties, have been shown to increase sensitivity to HMI and other analytes by providing binding sites on the electrode surface.[[10]] RGO therefore retains some of graphene’s excellent strength and conductivity while supporting defects uniquely useful for HMI detection.
There are multiple methods for synthesizing reduced graphene oxide, but electrochemical reduction has many advantages. Electrochemically reduced graphene oxide (ERGO) can be directly electrodeposited from a solution of graphene oxide (GO), which is hydrophilic and dissolves easily in polar solvents (e.g. water).[[9]] If the GO is reduced prior to deposition via chemical, thermal, or electrochemical means, it becomes hydrophobic and difficult to disperse in inorganic solvents, and is difficult to attach to the electrode surface without dropcasting The easily soluble precursor and simple, one-step fabrication method make electrochemical reduction is a very attractive technique.
ERGO has been used increasingly in literature to detect HMI electrochemically, often combined with other nanostructural modifications to enhance electrode surface area: Lee et al. combined one-step cyclic voltammetry (CV)-reduced ERGO with deposition of in situ bismuth film to detect Zn, Pb, and Cd ions[[11]] and Ping et al. used a one-step single-applied-potential method combined with in situ Bi to detect Pb and Cd ions.[[12]] Two-step ERGO, where GO is dropcast on a surface and then reduced, is also used and is commonly chosen due to its simplicity. However, it results in a thick film with an irregular distribution due to evaporation.[[9]] Furthermore, often in two-step ERGO only the top few layers of the deposition are reduced, leaving an insulating layer of GO next to the substrate.[[13]] To our knowledge, little work has been done developing a simple sensor using exclusively one-step ERGO.
This paper presents a simple, one-step CV method for the fabrication of unmodified ERGO sensors on GCE. The sensors were used to detect Pb2+ and Cd2+ down to nanomolar concentrations and were evaluated in the presence of other interfering metal ions. GO was verified by X-ray diffraction (XRD), UV–Vis spectrophotometry, and X-ray photoelectron spectroscopy (XPS); ERGO was verified by XPS and further characterized by electrochemical impedance spectroscopy (EIS).
Materials and methods
Reagents and apparatus
Sodium acetate trihydrate and cadmium chloride were purchased from Fisher (Fairlawn, NJ USA). GO precursor (graphene oxide 4–10% edge oxidized) and acetic acid were purchased from Sigma (St. Louis, MI USA). Lead (II) nitrate was purchased from Alfa Aesar (Ward Hill, MA USA). Ag/AgCl reference electrodes, Pt wire counter electrodes, GCE working electrodes, and potentiostats were purchased from CH Instruments, Inc. (Austin, TX USA). XRD spectra were taken on a Siemens D500 x-ray diffractometer. XPS spectra were taken on a Kratos Analytical AMICUS instrument. UV–Vis spectra were acquired on a Perkin Elmer Lambda 25 instrument.
Graphene oxide synthesis
Graphene oxide was synthesized via the Hummers method as reported in the literature.[[14,15,16]] To summarize: GO precursor was mixed with H2SO4 and KMnO4 and stirred at 35°C for 6 h. Next, the solution was diluted with deionized (DI) water and stirred for 2 h at room temperature. Finally, H2O2 was added to stop the reaction. The GO was washed with 10% HCl solution once and DI water twice and then ultrasonicated for one hour to exfoliate. The solution was then centrifuged at low speed for 5 min to precipitate the unexfoliated GO. The supernatant was dried in the oven at 60°C. The product was weighed and then resuspended in DI water for storage.
ERGO-GCE fabrication
GCE were cleaned with alumina slurry polishing and sonication, followed by cyclic voltammetry in 0.1 M sodium acetate buffer between 1 and − 1 V at a scan rate of 100 mV/s until a stable voltammogram was obtained. The cleaned GCE was utilized as the working electrode in an electrochemical cell containing a solution of 0.1 mg/mL GO suspended in DI water. An optimal number of voltammetric cycles were performed (− 1 V to 1 V at a scan rate of 50 mV/s) to directly reduce the GO onto the GCE surface.[[8, 9]] The ERGO-GCE was rinsed in DI water and allowed to air dry before use.
Cd2+ and Pb2+ detection
All electrochemical experiments were performed on the CHI 6051E/920D electrochemical instrument. Relevant concentrations of HMI precursor compounds were diluted with 0.1 M sodium acetate buffer solution (ABS) at pH 5, and thus all molarities refer to concentrations of the precursor compound. HMI were preconcentrated onto the ERGO-GCE at − 1.2 V for 180 s with stirring. Both pH values and deposition potential and time were chosen after a careful consideration of the relevant literature; − 1.2 V is applicable to any variety of HMI with a less negative oxidation potential,[[17, 18]] and pH 5 is both an exceptionally common value in literature and acidic enough to facilitate dissolved HMI without using concentrated acids.[[11, 17, 19]] After a quiescent time of 10 s, the HMI were stripped using differential pulse anodic stripping voltammetry (DPASV), where the step size was 4 mV, the amplitude was 0.1 V, the pulse width was 0.1 s, the sample width was 3 ms, the pulse period was 1 s, and the sensitivity was 0.001 A/V. The electrode surface was cleaned after each measurement by applying a potential of 0.3 V for at least 180 s with stirring.
Results and discussion
Graphene oxide characterization
Synthesized GO dried on an indium-tin oxide (ITO) glass slide was analyzed using XRD [Fig. 1(a)]. Well-defined peaks matched with reported values for the (001) plane of GO at 2θ = 9.1°,[[20]] with the small peak at 2θ = 27.0° belonging to either residual graphite (JCPDS 41-1487) or SnO2, and the remaining peaks corresponding to the underlying SnO2 (JCPDS 41-1445). UV–Vis spectra of GO dissolved in DI water were also obtained [Fig. 1(b)]. These spectra show a peak around 230 nm with a shoulder at 305 nm, which further confirms that the product is GO.[[21]] XRD spectra for ERGO on tin-oxide showed only values associated with SnO2 due to the thinness of the deposited layer, which is confirmed by published literature that indicates that RGO exhibits no diffraction peak when compared with GO, or else a very low, broad diffraction peak around 2θ = 23.5°.[[18, 20]] The XRD spectra for 12cyc ERGO on SnO2 can be found in Figure S1 of the Supporting Information.

(a) XRD spectra of GO dried on an ITO glass slide; (b) UV–Vis spectra of GO dissolved in DI water; XPS spectra of GO and ERGO, characteristic energy 1253.6 eV, (c) full survey (acquisition time 0.2 s) and (d) C 1 s (acquisition time 2 s for ERGO, 1 s for GO).
Electrochemically reduced graphene oxide characterization
XPS was chosen to verify the electrochemical reduction of GO. ERGO was deposited on a fluorine-doped tin oxide (FTO) glass slide using 15 voltammetric CV cycles with the parameters described above. This value was chosen to generate a film thick enough to be detected by XPS. Some GO solution was also dropcast on FTO. The resulting C 1 s spectra [Fig. 1(d)] indicate that the ERGO sample exhibited sp2-hybridized carbon–carbon bonds, indicative of a graphenic lattice, whereas the GO sample had two peaks due to the presence of carbon–oxygen bonds.[[18, 20]] Thus, reduction of GO and restoration of the graphenic lattice was confirmed by XPS. However, either due to incomplete coverage of the ERGO sample, or because the ERGO layer is < 10 nM, O 1 s spectra were also detected from the underlying SnO2 substrate, as seen in the full survey shown in Fig. 1(c). Thus while the C/O ratio was determined to be 7:3 for GO, the C/O ratio for ERGO could not be determined accurately.
Electrochemical characterization of modified electrode
EIS examinations of bare GCE, 4-cycle ERGO-GCE (4cyc), and 12-cycle ERGO-GCE (12cyc) revealed Nyquist plots (Fig. 2) that exhibit Faradaic mixed diffusion- and kinetic-controlled behavior when interacting with 5 mM redox probe K3[Fe(CN)6] in 0.1 M KCl.[[22, 23]] The data was fitted to a simplified Randles cell, excluding the Warburg region, to determine a charge transfer resistance (Rct) of 112.4 Ω ± 2.2 Ω for bare GCE, 107.4 Ω ± 2.4 Ω for 4cyc-ERGO, and 107.7 Ω ± 2.3 Ω for 12cyc-ERGO. Thus there is a ~5 Ω decrease in Rct upon deposition of ERGO on the glassy carbon surface. At lower frequencies (high Z′) behavior of the thicker 12cyc film diverges further from its bare and 4cyc counterparts in terms of the second time constant for the large semicircle that begins to emerge for all three samples (which is indicative of a finite-length diffusion layer.[[23]]) Thus 12cyc and 4cyc ERGO are electrochemically differentiable from the bare GCE substrate, and 12cyc is differentiable from 4cyc.

EIS Nyquist plots of bare GCE and GCE modified with 4cyc ERGO and 12cyc ERGO. Full frequency range was 10 Hz to 1 MHz, with applied oscillation potential of 10 mV, starting at 0 V. Inset: close-up of higher frequencies.
Optimization of deposition parameters
The number of CV cycles that control ERGO deposition had to be optimized for each analyte. Cycle number was varied from 3 to 18, and the parameters that led to the highest current in DPASV while detecting a set concentration of the analyte. The 12cyc and 4cyc parameters both performed well for Pb2+, and the 3cyc and 12cyc parameters performed well for Cd2+. 12cyc was used going forward due to its higher sensitivity toward Cd2+ and greater robustness. The results of the cycle number experiments can be found in more detail in Figure S2 of the Supporting Information.
Differential pulse voltammetry
DPASV of heavy metals follows a distinct pattern on ERGO-GCE, with up to three oxidation peaks being present in the voltammogram: a peak around – 0.2 V that is attributed to high-energy metal-ERGO bonds, a peak at the primary oxidation voltage for each metal (around – 0.65 V for Pb2+ and − 0.9 V for Cd2+) attributed to metal–metal bonds, and a shoulder at a slightly more positive potential than the primary oxidation peak attributed to medium energy metal-ERGO bonds in competition with metal–metal bonds.[[24]] These peaks occur regularly during DPASV detection of HMI. With increasing HMI concentration, the “shoulder” (– 0.6 V for Pb2+, – 0.75 V for Cd2+) plateaus as preferred ERGO binding sites are filled, and the primary metal–metal peak begins to grow. Similarly, HMI deposition can be modeled as a “two-plate” system, in which a primary layer of metal forms on the electrode surface and the subsequent layer forms on top of that.[[25]]
Limit of detection (LOD) for 12cyc ERGO-GCE was determined experimentally as 5 nM for Pb2+ and 75 nM for Cd2+ (S/N > 3), which is 1.04 µg/L for lead and 8.67 µg/L for cadmium in terms of the mass of the ions themselves. Calibration experiments were also performed. The calibration curves for both analytes are shown in Fig. 3. Two distinct calibration regions were found for each metal,[[18, 26]] with a transition from “low” to “high” around 2.0 µM. The regression equations are included below (1–4) for “low” and “high” concentration regions for both Pb2+ and Cd2+.

(a) DPVs and (b) calibration curve for Cd2+ (n = 3); (c) DPVs and (d) calibration curve for Pb2+ (n = 3).
Parts (a) and (c) in Fig. 3 clearly demonstrate the leftward peak shift with increasing metal concentration, with effects on peak magnitude that are difficult to predict. Metal deposition and detection is a complex system, as the act of depositing metal on the electrode surface changes the surface properties slightly.[[25]] The shallower sensitivity in the range of 0.5–2.0 µM correlates with primary peak growth around − 0.65 V, with the peak at − 0.6 V remaining as a shoulder. The function of the ERGO deposition on the GCE surface is to facilitate the initial adsorption of HMI at preferred binding sites.[[27]]
Simultaneous detection of Pb and Cd ions reveals a large separation between oxidation peaks and no interference, shown in Fig. 4. The Pb2+ signal is consistently much larger than the Cd2+ signal at the same concentration, and more sensitive to change in analyte concentration, which may be an effect of the Pb ion’s less negative oxidation voltage. When the concentration of Cd2+ was varied from 2 to 8 µM in increments of 2 µM while the concentration of Pb2+ was held constant at 2 µM, the magnitude of the Cd2+ peak increased with increasing concentration while the Pb2+ peak magnitude held mostly constant [Fig. 4(a)]. The relative standard error (RSE) for the Pb2+ peak was 4.4%. Likewise, when the concentration of Cd2+ was held constant at 2 µM and the concentration of Pb2+ was varied from 2 to 3.5 µM in increments of 0.5 µM, the magnitude of the Cd2+ peak held constant while the magnitude of the Pb2+ peak increased regularly [Fig. 4(b)]. The RSE for the Cd2+ peak was 9.0%. Thus we can conclude that the Cd and Pb ions do not interfere with each other during detection.

DPV plots of simultaneous detection of Pb and Cd on 12cyc ERGO-GCE: (a) holding the concentration of Pb2+ constant at 2 µM and increasing Cd2+ concentration, and (b) holding the concentration of Cd2+ constant at 2 µM while increasing Pb2+ concentration.
Stability and reproducibility
A set of stability and reproducibility tests was performed using 4cyc ERGO-GCE and 1 µM Pb2+. Average relative standard deviation (RSD) across three separate samples was 5.4%, while average standard deviation across the same sample tested three times over a period of 26 days was 6.4%. For 12cyc ERGO-GCE, three consecutive tests on the same sample for 1 µM Pb2+ yielded an RSD value of 4.5%. These excellent reproducibility numbers indicate even thin 4cyc-ERGO-GCE sensors are both stable and reliable. Although the sensors are intended for single use, these tests prove that they can be reused when necessary.
Interference
Cd and Pb ions at a concentration of 2 µM were tested simultaneously via DPASV in the presence of 200 µM zinc (Zn2+) and iron (III) (Fe3+). These two ions were chosen as they often serve as co-contaminants in the environment.[[5]] No deleterious effect on the Cd and Pb ion signals were found in the presence of Zn and Fe ions when compared to an un-spiked control sample containing only Cd and Pb ions. A small Zn2+ peak could be discerned at an oxidation potential near − 1.1 V. Variation was found to be within the standard error of the un-spiked sample, as shown in Figure S3. From this we can conclude that the ERGO-GCE sensor functions as intended even in the presence of other metals. Additionally, a sample volume of 5 mL of tap water collected from the city of Ames, IA was spiked with 5 µM each of Cd2+ and Pb2+. The sample was diluted with ABS and detected using the ERGO-GCE sensor. Recoveries are shown in Table S1, and DPV scans are shown in Fig. S4. Recovery for Pb2+ was 93.5%, but recovery for Cd2+ was 44.1%. Additionally, the Pb2+ peak had a larger and more separated shoulder region. This is attributed to complexation with additive ions (e.g. fluoride) present in the tap water.
Comparison to literature
Our sensor compares well to other sensors reported in literature, as shown in Table S2, all of which are in the nanomolar range. Although our LOD for Cd2+ is large compared to many others, it was achieved without the aid of any other nanomaterial surface modifications or bismuth fused alloys.
Conclusion
In conclusion, this paper demonstrates a simple, robust ERGO sensor capable of achieving nanomolar detection limits of Cd and Pb HMI without the use of additional nanostructure modifications or in situ fused alloys. The ERGO was fabricated using a straightforward one-step CV method at room temperature and verified with XPS. The sensor had a linear relationship in a useful detection range for both Pb and Cd ions, with LOD of 5 nM for Pb2+ and 75 nM for Cd2+. Furthermore, the sensor could detect both ions simultaneously with no interference, was robust and reliable with repeated use, and continued to function normally in the presence of co-contaminant Fe and Zn ions. Thus our sensor material exhibits great promise for future development of sensitive and inexpensive sensors for HMI quantification in the environment.
Data availability
Data will be made available upon reasonable request.
Code availability
Not applicable.
References
P.B. Tchounwou, C.G. Yedjou, A.K. Patlolla, D.J. Sutton, Heavy metals toxicity and the environment. EXS 101, 133–164 (2012). https://doi.org/10.1007/978-3-7643-8340-4_6
V.D. Zheljazkov, L.E. Craker, B. Xing, Effects of Cd, Pb, and Cu on growth and essential oil contents in dill, peppermint, and basil. Environ. Exp. Bot. 58, 9–16 (2006). https://doi.org/10.1016/j.envexpbot.2005.06.008
W. Tan, Q. Gao, C. Deng, Y. Wang, W.-Y. Lee, J.A. Hernandez-Viezcas, J.R. Peralta-Videa, J.L. Gardea-Torresdey, Foliar exposure of Cu(OH)2 nanopesticide to basil (Ocimum basilicum): variety-dependent copper translocation and biochemical responses. J. Agric. Food Chem. 66, 3358–3366 (2018). https://doi.org/10.1021/acs.jafc.8b00339
D.S. Kacholi, M. Sahu, Levels and health risk assessment of heavy metals in soil, water, and vegetables of Dar es Salaam, Tanzania. J. Chem. 2018, e1402674 (2018). https://doi.org/10.1155/2018/1402674
L. Rodriguez-Freire, S. Avasarala, A.-M.S. Ali, D. Agnew, J.H. Hoover, K. Artyushkova, D.E. Latta, E.J. Peterson, J. Lewis, L.J. Crossey, A.J. Brearley, J.M. Cerrato, Post gold king mine spill investigation of metal stability in water and sediments of the animas river watershed. Environ. Sci. Technol. 50, 11539–11548 (2016). https://doi.org/10.1021/acs.est.6b03092
S. Lee, S. Bong, J. Ha, M. Kwak, S.-K. Park, Y. Piao, Electrochemical deposition of bismuth on activated graphene-nafion composite for anodic stripping voltammetric determination of trace heavy metals. Sens. Actuators B Chem. 215, 62–69 (2015). https://doi.org/10.1016/j.snb.2015.03.032
R. De Penning, N. Monzon, S. Padalkar, Flexible zinc oxide-based biosensors for detection of multiple analytes. J. Mater. Res. 37, 2942–2950 (2022). https://doi.org/10.1557/s43578-022-00693-0
W. Gao, The chemistry of graphene oxide, in Graphene Oxide. (Springer, Cham, 2015), pp.61–95. https://doi.org/10.1007/978-3-319-15500-5_3
S.Y. Toh, K.S. Loh, S.K. Kamarudin, W.R.W. Daud, Graphene production via electrochemical reduction of graphene oxide: synthesis and characterisation. Chem. Eng. J. 251, 422–434 (2014). https://doi.org/10.1016/j.cej.2014.04.004
W. Peng, H. Li, Y. Liu, S. Song, A review on heavy metal ions adsorption from water by graphene oxide and its composites. J. Mol. Liq. 230, 496–504 (2017). https://doi.org/10.1016/j.molliq.2017.01.064
S. Lee, S.-K. Park, E. Choi, Y. Piao, Voltammetric determination of trace heavy metals using an electrochemically deposited graphene/bismuth nanocomposite film-modified glassy carbon electrode. J. Electroanal. Chem. 766, 120–127 (2016). https://doi.org/10.1016/j.jelechem.2016.02.003
J. Ping, Y. Wang, J. Wu, Y. Ying, Development of an electrochemically reduced graphene oxide modified disposable bismuth film electrode and its application for stripping analysis of heavy metals in milk. Food Chem. 151, 65–71 (2014). https://doi.org/10.1016/j.foodchem.2013.11.026
S. Kumar Bikkarolla, P. Cumpson, P. Joseph, P. Papakonstantinou, Oxygen reduction reaction by electrochemically reduced graphene oxide. Faraday Discuss 173, 415–428 (2014). https://doi.org/10.1039/C4FD00088A
M.B. Gumpu, M. Veerapandian, U.M. Krishnan, J.B.B. Rayappan, Simultaneous electrochemical detection of Cd(II), Pb(II), AS(III) and Hg(II) ions using ruthenium(II)-textured graphene oxide nanocomposite. Talanta 162, 574–582 (2017). https://doi.org/10.1016/j.talanta.2016.10.076
Y. Shao, J. Wang, M. Engelhard, C. Wang, Y. Lin, Facile and controllable electrochemical reduction of graphene oxide and its applications. J. Mater. Chem. 20, 743–748 (2010). https://doi.org/10.1039/B917975E
F.X. Hu, J.L. Xie, S.J. Bao, L. Yu, C.M. Li, Shape-controlled ceria-reduced graphene oxide nanocomposites toward high-sensitive in situ detection of nitric oxide. Biosens. Bioelectron. 70, 310–317 (2015). https://doi.org/10.1016/j.bios.2015.03.056
Y. Zuo, J. Xu, F. Jiang, X. Duan, L. Lu, G. Ye, C. Li, Y. Yu, Utilization of AuNPs dotted S-doped carbon nanoflakes as electrochemical sensing platform for simultaneous determination of Cu (II) and Hg (II). J. Electroanal. Chem. 794, 71–77 (2017). https://doi.org/10.1016/j.jelechem.2017.04.002
Z. Guo, D. Li, X. Luo, Y. Li, Q.-N. Zhao, M. Li, Y. Zhao, T. Sun, C. Ma, Simultaneous determination of trace Cd(II), Pb(II) and Cu(II) by differential pulse anodic stripping voltammetry using a reduced graphene oxide-chitosan/poly-l-lysine nanocomposite modified glassy carbon electrode. J. Colloid Interface Sci. 490, 11–22 (2017). https://doi.org/10.1016/j.jcis.2016.11.006
P.S. Adarakatti, V.K. Gangaiah, C.E. Banks, A. Siddaramanna, One-pot synthesis of Mn3O4/graphitic carbon nanoparticles for simultaneous nanomolar detection of Pb(II), Cd(II) and Hg(II). J. Mater. Sci. 53, 4961–4973 (2018). https://doi.org/10.1007/s10853-017-1896-6
R. Al-Gaashani, A. Najjar, Y. Zakaria, S. Mansour, M.A. Atieh, XPS and structural studies of high quality graphene oxide and reduced graphene oxide prepared by different chemical oxidation methods. Ceram. Int. 45, 14439–14448 (2019). https://doi.org/10.1016/j.ceramint.2019.04.165
S. Xu, L. Yong, P. Wu, One-pot, green, rapid synthesis of flowerlike gold nanoparticles/reduced graphene oxide composite with regenerated silk fibroin as efficient oxygen reduction electrocatalysts. ACS Appl. Mater. Interfaces 5, 654–662 (2013). https://doi.org/10.1021/am302076x
J. Muñoz, R. Montes, M. Baeza, Trends in electrochemical impedance spectroscopy involving nanocomposite transducers: characterization, architecture surface and bio-sensing. Trends Anal. Chem. 97, 201–215 (2017). https://doi.org/10.1016/j.trac.2017.08.012
E. Casero, A.M. Parra-Alfambra, M.D. Petit-Domínguez, F. Pariente, E. Lorenzo, C. Alonso, Differentiation between graphene oxide and reduced graphene by electrochemical impedance spectroscopy (EIS). Electrochem. Commun. 20, 63–66 (2012). https://doi.org/10.1016/j.elecom.2012.04.002
K.C. Honeychurch, J.P. Hart, D.C. Cowell, Voltammetric behavior and trace determination of lead at a mercury-free screen-printed carbon electrode. Electroanalysis 12, 171–177 (2000). https://doi.org/10.1002/(SICI)1521-4109(200002)12:3%3c171::AID-ELAN171%3e3.0.CO;2-Q
P.M. Skitał, P.T. Sanecki, D. Saletnik, The investigation and modeling of two metals codeposition process. J. Electroanal. Chem. 778, 87–97 (2016). https://doi.org/10.1016/j.jelechem.2016.08.002
H. Xing, J. Xu, X. Zhu, X. Duan, L. Lu, Y. Zuo, Y. Zhang, W. Wang, A new electrochemical sensor based on carboimidazole grafted reduced graphene oxide for simultaneous detection of Hg2+ and Pb2+. J. Electroanal. Chem. 782, 250–255 (2016). https://doi.org/10.1016/j.jelechem.2016.10.043
P.M. Skitał, P.T. Sanecki, D. Saletnik, The modeling of simultaneous three metals codeposition investigated by cyclic voltammetry. J. Electroanal. Chem. 878, 114698 (2020). https://doi.org/10.1016/j.jelechem.2020.114698
Acknowledgments
The authors would like to thank Dapeng Jing from the Materials Analysis Research Laboratory for his help gathering and interpreting XPS data.
Funding
The authors would like to thank the Catron Fellowship, Brown Graduate Fellowship, and Iowa State University Startup Fund for their funding support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
De Penning, R., Padalkar, S. Heavy metal sensing in water using electrochemically reduced graphene oxide. MRS Communications 13, 531–537 (2023). https://doi.org/10.1557/s43579-023-00369-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43579-023-00369-8