
Abstract
The current work reports the biocompatibility and mechanical stability of enstatite and forsterite bioceramics prepared by sol–gel combustion method. XRD results conferred that enstatite and forsterite phase formation take place at 1000 °C and 900 °C respectively. TEM micrographs indicated the particle size of enstatite in the micron range while forsterite is in the range of 100–200 nm. The FT-IR spectra of forsterite after biomineralization revealed the presence of phosphate and carbonate groups shows apatite deposition ability of forsterite. The slow degradation and better apatite deposition of forsterite resulted in ten folds greater compressive strength than enstatite. Both the bioceramics have shown a remarkable impact on inhibiting the growth of clinical pathogens at a very low concentration. The good hBMSCs attachment and significant proliferation revealed the cytocompatibility of enstatite and forsterite. These observations suggested that magnesium silicate bioceramics can be explored for load-bearing applications, maxillofacial reconstruction and septic arthritis.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the advancements in orthopedic technologies for hard tissue regeneration, an increase in the demand for biomaterials is observed. The criteria for fabricating an ideal scaffold applicable for bone regeneration not only demand good biocompatibility but sufficient structural support to the regenerating tissues during degradation and remodelling [1]. Magnesium-containing silicate bioceramics are new hope in this field. Although substantial work has been carried out on calcium silicates as well as calcium phosphates, these bioceramics do not fulfill the complete requirements for hard tissue replacement. The lack of proper vascularization and insufficient mechanical stability limits their load bearing and biomedical applications [2]. Magnesium containing silicate bioceramics are found to be bioactive, possess slow degradation, and exhibit good mechanical properties as compared to calcium silicates as well as calcium phosphates [3]. Moreover, the release of magnesium and silicon ions stimulates the proliferation and adhesion of osteoblast cells [4, 5]. Hence, extensive research has been conducted by various research groups towards exploring magnesium silicate bioceramic for hard tissue regeneration.
Forsterite (Mg2SiO4) and enstatite (MgSiO3) are the most common magnesium silicate bioceramics studied for bone tissue regeneration. Moreover, enstatite exists in three polymorphic forms (orthoenstatite, clinoenstatite, protoenstatite) [6]. Earlier, Diba et al., summarized the synthesis, sintering, mechanical properties, degradation, in-vitro as well as in-vivo biological behavior of magnesium-containing bioactive silicate ceramics [2]. It was reported that the presence of magnesium ions in forsterite suppresses the apatite deposition ability as well as results in slow degradation [7, 8]. Naghiu et al., reported nano forsterite can precipitate bone-like apatite over its surface within one week of immersion and thus nano forsterite offers better apatite deposition ability and degradation rate than micron size [9, 10]. Previously, forsterite has been indicated as a potential antibacterial bioceramic with enhanced mechanical stability for load-bearing applications [11]. Recently, biomineralization, dissolution, and cellular behavior of forsterite prepared from biowaste were investigated [12]. Biowaste-derived forsterite was found to have compatibility with mammalian blood cells. Gorea et al., emphasized the ability of porous forsterite to stimulate and promote the adhesion, proliferation, and growth of fibroblast cells and concluded it as a potential bone substitute [13]. The interaction between nano forsterite and different cell lines such as U20S human osteoblast cells and osteoblast G292 cells revealed its excellent cytocompatibility [9, 14]. Lakshmi et al., reported forsterite/wollastonite composites resulted in better apatite deposition over their surface [15]. Moreover, forsterite/diopside composites were fabricated to develop mechanically stable and biocompatible scaffolds for bone tissue regeneration. This study revealed excellent cell viability, good antibacterial activity, slow degradation, and remarkable mechanical strength was achieved as the content of forsterite was increased in the composites [16]. Nabiyouni et al., reviewed the potential of magnesium-containing bioceramics (MgO–P2O5, CaO–MgO–P2O5, MgO–SiO2) for tissue engineering applications [3]. It was suggested that the in-vivo studies of these bioceramics need to be performed in large animal models to predict their orthopedic applications. In addition, this approach will assist to understand the bone regeneration ability in humans.
Venkatraman and Sasikumar suggested that the biomineralization ability of enstatite can be enhanced by preparing composites with wollastonite [17]. Earlier, enstatite bioceramic was suggested as a promising candidate for bone and dental restorations as it has supported the deposition of mineralized bone tissue [6]. A literature study reveals that clinoenstatite was used as a coating material and exhibits cytocompatibility and immune-modulatory effects with the release of Mg and Si ions. It was also suggested that clinoenstatite coatings improved osteointegration with host bone during in-vivo studies [18]. In another study, clinoenstatite was unable to induce bone-like apatite precipitation over its surface but the release of ions from clinoenstatite stimulated the proliferation of L929 mouse fibroblast cells [19]. Moreover, the adhesion and proliferation of murine embryonic mesenchymal stem cells (MEMST) on clinoenstatite was more as compared to the hydroxyapatite bioceramic. In the case of protoenstatite, only one report has been published which indicated its biomineralization ability in physiological body fluid [20].
The major problem associated with forsterite and enstatite is that their pure phase formation occurs at an elevated temperature when prepared by traditional methods. Various synthetic procedures have been employed for the preparation of pure forsterite such as citrate–nitrate method [21], microwave sintering [22], sol–gel [23], mechanical activation process [24], co-precipitation [25], solid-state method [26],and sol–gel combustion method [10]. It had been reported that the phase purity of forsterite was achieved at high temperatures (1200–1500 °C) by the majority of these preparatory routes. Surfactant-assisted foam technique was utilized to prepare enstatite from talc at 1200 °C with protoenstatite, cristobalite, and diopside as impurities [27]. All the three forms of MgSiO3 were observed when the raw materials were ball milled and calcined at 1200 °C [28]. The geo-polymer technique was used to prepare enstatite at 1100 °C with forsterite as a secondary phase [29]. Pure enstatite was achieved at 1300 °C through the sol–gel technique [30].
Literature studies have indicated that septic arthritis, osteomyelitis, and prosthetic joint infections are the major diseases associated with bone, which involve the inflammatory destruction of joints and bone due to bacterial infections [31]. It is estimated that the total number of people above 50 years of age affected by bone diseases will be doubled in the current decade [32]. Nearly 95% of bone diseases are occurring due to four bacterial strains such as Staphylococcus aureus (S. aureus), Staphylococcus epidermidis (S. epidermidis), Escherichia coli (E. coli) and Pseudomonas aeruginosa (P. aeruginosa). Since Staphylococcus strains produce bio-films which could not be resisted by antibiotic and the immune system. Further, adhesion of 18–20 bio-film inhibiting bacteria is required to avoid implant infections [33]. The bacterial infections occurred during surgery affects neighbor cells and tissues that lead to implant failure or even complete removal of the infected organ [34]. These issues can be overwhelmed by designing implantable biomaterial having both antibacterial activity as well as cytocompatibility to guide and promote bone regeneration.
The objective of the present work is to synthesize forsterite and enstatite at low temperature by an economically efficient sol–gel assisted combustion route and optimize the parameters to achieve the phase purity. Various physicochemical characterization studies were carried out for analyzing the influence of fuel-citric acid on crystallization temperature, surface morphology, and particle size. Furthermore, the bactericidal activity of pure forsterite and enstatite against gram-positive and gram-negative bacteria as well as biocompatibility against hBMSCs were investigated and compared. The influence of degradation and biomineralization on mechanical stability concerning compressive strength and young’s modulus were also examined.
Results
Synthesis of enstatite and forsterite
Studies have indicated the dual role of fuel in the sol–gel combustion method [33]. Citric acid used as a fuel in the present work formed metal-citrate complex which avoided the precipitation of metal ions in the reaction mixture. Nitric acid acted as a catalyst by increasing the rate of the hydrolysis reaction. Initially, tetraethyl orthosilicate reacts with water (hydrolysis) to form silanol and ethanol as per Eq. 1. Silanol undergoes condensation reaction with the other neighboring silanol molecules and precedes the polycondensation with metal-citrate complex leading to the formation of aerogel.
Moreover, citric acid (fuel) present in the dried gel acted as a reducing agent whereas nitrates as an oxidizing agent during the combustion reaction. Thus, citric acid and nitrate reacted under the influence of heat and initiated a self-propagating combustion process with the evolution of carbon dioxide and water vapour. It has been reported that the amount of heat energy liberated during combustion reaction depends upon the fuel and its molar ratio with metal nitrates [35]. The release of gases during the combustion process induces porosity in the precursor and the heat generated during redox reaction causes crystallization of the product. To achieve the phase purity of magnesium silicates, the decomposed precursors were calcined at different temperatures for various time intervals. The overall reaction involved during the synthesis of enstatite and forsterite is shown in Eq. 2 and Eq. 3.
The enstatite was calcined at 1000 °C and the forsterite was calcined at 900 °C to get the pure phase with the elimination of secondary phases.
Characterization of enstatite and forsterite
Thermal and XRD analysis
The thermo gravimetric and differential thermal analysis (TG–DTA) of enstatite is shown in Fig. 1a. The thermal analysis indicates three critical stages: the initial weight loss of 44% is attributed to the removal of moisture content which is accompanied by a broad endothermic peak at 110 °C in DTA. Later, a weight loss of about 18% was noticed due to exothermic peaks at 211 °C, and 262 °C. This is assigned to the decomposition of fuel during the combustion reaction [10]. Finally, the elimination of organic residues leads to a weight loss of about 20%. An exothermic peak at 408 °C is attributed to the formation of periclase whereas the broad exothermic peak around 550 °C indicates the decomposition of carbonaceous residues [21]. Moreover, the exothermic peak at 920 °C shows the crystallization temperature of the enstatite.

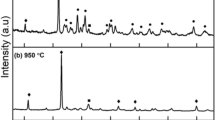
TGA/DTA thermogram and XRD patterns of (a, c) enstatite (b, d) forsterite.
The thermal behaviour of the forsterite precursor (Fig. 1b) was observed to have similar weight loss patterns as that of enstatite. The broad endothermic peak at 120 °C indicates the dehydration of precursor leading to 35% of weight loss. The release of water vapour and carbon dioxide during the combustion process resulted in the appearance of dual exothermic peaks at 230 °C to 240 °C resulted in a weight loss of nearly 22.5%. The small exothermic peak at 400 °C is attributed to the formation of periclase. The final weight loss of ~ 15% at 570 °C is due to the elimination of organic moieties [21]. Moreover, the crystallization of forsterite was observed at 845 °C [10, 21].
The X-ray diffraction pattern of enstatite calcined at 1000 °C is illustrated in Fig. 1c. According to our previous findings on enstatite [17], the phase evolution occurs as follows: after combustion of the precursor at 400 °C, cristobalite and periclase phases were observed. Since the dried gel precursor was hygroscopic, water molecules were absorbed from the environment. When the dried gel powder was subjected to the combustion at 400 °C, the exothermicity was not sufficient to achieve the enstatite phase. When the calcination temperature was increased to 600 °C for 6 h, the appearance of forsterite was noticed as a major phase and enstatite was observed as a secondary phase. It might be due to the reaction between two periclase (MgO) molecules with a cristobalite (SiO2) molecule. To achieve single phasic enstatite, the impurity (forsterite) was disintegrated by the action of heat treatment. As the calcination temperature was raised to 1000 °C for 12 h, enstatite phase formation was achieved without any secondary phase impurities. The enstatite pattern obtained after calcination at 1000 °C for 12 h was indexed with standard JCPDS file no: 96–900-6340 (Fig. 1c). The lattice parameter calculated from the powder XRD was found to be a = 9.25 Å, b = 8.74 Å, c = 5.31 Å. The average crystallite size of the enstatite was found to be 44–48 nm, which was calculated by using the Scherrer equation (D = kλ/β Cos θ).
Figure 1d shows the XRD pattern of forsterite calcined at 900 °C. As per the results obtained from our previous findings on forsterite [17], it was noticed that the gel decomposed at 400 °C shows the formation of the forsterite phase along with periclase and cristobalite phases as impurities. The presence of enstatite was noticed even after calcination at 700 °C for 6 h. The complete elimination of secondary phases from the forsterite pattern was observed after calcination at 900 °C for 6 h. The XRD pattern of single phasic forsterite calcined at 900 °C was found to match with standard JCPDS data card: 96-900-0320. It was observed that it possesses an orthorhombic crystal system with lattice parameters a = 4.76 Å, b = 10.2 Å and c = 5.98 Å (Fig. 1d). Further, with the help of the Scherrer equation, the crystallite size of forsterite was calculated as 30–34 nm.
Hence, in the current work calcination temperature was reduced without using mechanical activation, ball milling or surfactants to achieve pure forsterite and enstatite.
Morphological investigation
The surface morphology, elemental composition and TEM/SAED patterns of enstatite are shown in Fig. 2. The SEM micrographs of enstatite calcined at 1000 °C for 12 h (Fig. 2a and b) showed a scattering of agglomerated particles over the surface having an irregular shape. The irregular shape with different size distribution was due to the multistep decomposition [36]. The EDAX spectrum (Fig. 2c) shows the presence of Mg, Si and O peaks which confirms the elemental composition of the enstatite. The TEM/SAED pattern of enstatite resulting from the sol–gel assisted combustion reaction is shown in Fig. 2d–f. The bright-field TEM analysis shows the supporting evidence of agglomeration throughout the material. On the other hand, the SAED pattern reveals the crystallinity with high order associated with the material. Since the particles are agglomerated due to high calcination temperature, the particle size observed from TEM analysis is in the micron range.

SEM (a, b)/EDAX (c) and TEM (d, e)/SAED (f) Micrographs of Enstatite.
The SEM/EDAX and TEM/SAED pattern of pure forsterite are shown in Fig. 3. SEM micrographs (Fig. 3a and b) revealed the presence of rough texture with scattered particles all over the surface. Moreover, the agglomeration of particles might have resulted due to multi-steps decomposition [37]. The chemical composition of forsterite has confirmed the presence of all elements’ peak in the EDAX spectra (Fig. 3c). The TEM micrograph of forsterite indicated the size of the particles in the range of 100–200 nm. The d-spacing value was found to be 0.297 nm from the TEM image is in good agreement with the XRD results. The SAED pattern revealed high crystallinity order of the forsterite sample. However, the high calcination temperature imparts the increase in particle size of the forsterite because of the grain growth phenomenon.

SEM (a, b)/EDAX (c) and TEM (d, e)/SAED (f) Micrographs of Forsterite.
In-vitro studies of enstatite and forsterite
Apatite formation ability
The biomineralization ability of enstatite was studied by immersing in SBF as shown in Fig. 4. The XRD pattern shows (Fig. 4a) that there was no sign of apatite deposition over the surface of the enstatite even after 28 days of immersion. However, the minor amorphous phase was noticed along with the enstatite phase, which might be due to the initiation of biomineralization. The absence of carbonate and phosphate functional groups in the FT-IR spectra (Fig. 4b) after biomineralization studies indicated poor apatite deposition behaviour of enstatite in the physiological environment. Thus, the absorption bands obtained in the FT-IR spectrum of enstatite after soaking in SBF were similar to that of pure enstatite. SEM micrograph revealed negligible deposition over the surface of the scaffold and the presence of Ca and P peaks were not detected in EDAX spectra which confirms the slow biomineralization potential of enstatite.

XRD (a), FT-IR (b) and SEM/EDAX (c) of enstatite immersed in SBF.
The XRD pattern (Fig. 5a) of forsterite showed no significant apatite peaks after immersion in SBF. An increase in the appearance of an amorphous phase was noticed over the immersed surface from 21st to 28th day. This might be due to the initiation of biomineralization on the surface of the forsterite scaffold. To confirm it, the immersed surface was characterized by FT-IR and SEM/EDAX analysis. The FT-IR (Fig. 5b) spectrum showed the existence of phosphate and carbonate functional groups. The absorption band at around 1400 cm−1 is assigned to the CO32− group, whereas the peaks between 1000 cm−1 to 1100 cm−1 and 500 cm−1 to 600 cm−1 are associated with PO43− groups [38]. These observations revealed that the amorphous phase noticed in the XRD pattern could be hydroxyapatite. The SEM micrographs (Fig. 5c) illustrated the presence of small particles deposited over the surface of the scaffold. The existence of Ca and P peaks in the EDAX spectrum confirmed the deposition of apatite on the surface of forsterite. This finding indicates that the forsterite can induce HAp at a slower rate. Even though both enstatite and forsterite are magnesium silicates, forsterite shows very low biomineralisation due to the presence of optimum level (30–45%) of SiO2 whereas enstatite possess restricted level (~ 60%) of SiO2 hence no biomineralization was observed. The onset of biomineralization starts on 21st day due to the leaching of Mg2+ ion from the forsterite matrix which delays the formation of stable apatite over the surface of silicate bioceramics [39].

XRD (a), FT-IR (b) and SEM/EDAX (c) of forsterite immersed in SBF.
Resorbability studies and mechanical strength evaluation
The resorbability plot with the weight gain/loss method of enstatite and forsterite is shown in Fig. 6a, b. Considering the simultaneous deposition of HAp and degradation of enstatite soaked in SBF, there is no considerable weight change observed during the first week of immersion. This weight observation indicates that there was neither degradation nor apatite deposition took place in the early days of immersion. A linear decrease in weight was found as the immersion time increased from 7 to 28th day due to the high degradation of the material into the physiological environment. Thus, the degradation of enstatite in SBF might have increased magnesium ion concentration in SBF which could have prevented the apatite deposition over its surface. Moreover, previous findings have suggested that high magnesium content in SBF suppresses the biomineralization over the surface of the scaffolds [7, 10].

Weight observation of enstatite (a) and forsterite (b) during in vitro bioactivity and c Comparative mechanical strength of enstatite and forsterite immersed in SBF.
The degradation behaviour of forsterite was found to be distinct from that of enstatite. In the case of forsterite, a continuous weight loss was observed till 14 days of immersion. When the samples were analyzed after two weeks, the change in weight was found to take place at a very slow rate. This can be correlated with the initiation of apatite deposition over the surface of forsterite which reduced the rate of degradation. Further, as the immersion time was increased from 21st to 28th day, a linear increase in weight was found. This observation revealed that the biomineralization dominates over the degradation of forsterite. During the immersion in SBF, degradation and biomineralization take place simultaneously. Degradation can lead to weight loss and decline in strength whereas biomineralization causes an increase in mechanical stability by gaining weight. Thus, the final weight change and strength of a material depend upon which mechanism occurs dominantly.
The mechanical property of forsterite and enstatite in terms of compressive strength and young’s modulus was studied as shown in Fig. 6c. The compressive strength of forsterite was found to be ten folds greater than enstatite. A similar trend was noticed in the case of young’s modulus, where forsterite bears the load about twenty times higher than enstatite. This variation between the two magnesium silicate materials is due to the degradation and apatite deposition. Chen et al., suggested that the mechanical properties of a material can be controlled or altered by adjusting the magnesium oxide content in the material [40]. Thus, the presence of more amount of magnesium oxide in forsterite (2MgO–SiO2) also led to superior mechanical strength over enstatite (MgO–SiO2).
Antibacterial activity assay
The results observed for inhibiting bacterial growth by the bioceramic samples are mentioned in Table 1. Both the magnesium silicate bioceramics showed excellent antimicrobial activity against Gram-positive test strain: S. aureus and S. epidermidis and Gram-negative test strain: E. coli and P. aeruginosa. The bactericidal property disrupts the cell functions such as cell differentiation, adhesion, and growth of bacteria, thereby inhibiting the biofilm formation [41].
Both the magnesium silicate biomaterials have the capability of clustering around the microorganisms and eventually rupture the cell membrane proteins due to biocompatibility property, thereby resulting in the leaching of genetic material and cellular protein content [26]. The rupture of the cell membrane was observed using a scanning electron microscope, as shown in SEM image Fig. 7. The SEM image of control S. aureus (Fig. 7a) showed a smooth surface compared to the post-experiment test strain: S. aureus with both biomaterials (Fig. 7b, c). These observations indicate magnesium silicate as a potential biomaterial for the treatment of bacterial infections such as osteomyelitis and septic arthritis.

SEM Micrographs post antibacterial assay against S. aureus with Control (a), Forsterite (b) and Enstatite (c).
In vitro biocompatibility
The interaction of enstatite and forsterite scaffold with hBMSCs was investigated and the cell attachment analysis was determined by using the SEM technique. The SEM micrograph images were taken on day 14 of the cell-scaffold culture duration and representative of three individual experiments (Kruskal–Wallis test for Alamar blue assay analysis: Cell seeded Enstatite and Forsterite scaffolds on Day 3, 7 and 14 Vs. Day 1 culture). Figure 8a and c confirmed that enstatite and forsterite scaffolds have provided a favourable environment for the cell attachment. This could be a reason due to an ideal chemical composition that supports cell surface adherent on a foreign body. This finding is an important validation to demonstrate the biocompatible feature of enstatite and forsterite.

SEM Micrographs post Viability of hBMSCs attached on Enstatite (a, b) and Forsterite (c, d).
Furthermore, Fig. 8b and d have shown the evidence that the cells started secreting extracellular matrix (ECM) like components surrounding the cell vicinity suggesting the cellular microenvironment preparation for its survival, proliferation, and differentiation [42]. This cell survival activity was further confirmed using Alamar blue cell viability/proliferation assay. The AB cell viability assay confirms that the enstatite and forsterite maintained the viability of hBMSCs throughout the culture duration (Fig. 9).

Alamar blue percentage of hBMSCs reduction on day 1, 3, 7 and 14.
Discussion
The overall biomineralization study indicated that both enstatite and forsterite did not have significant apatite deposition ability. Amongst both, forsterite showed better activity when compared to enstatite. Tavangarian et al., and Choudhary et al., have reported that nano-forsterite powder stimulates good bioactivity than micron-sized powders [10, 24]. Naghiu et al., reported that forsterite prepared at 1000 °C with micron-sized induced apatite deposition to a small extent even after 28 days of immersion in SBF [8]. The present result showed that bioactivity of forsterite with 100–200 nm in SBF is similar to the previous findings. Previous reports revealed that the presence of silica would induce apatite deposition at its lower compositional ratio in the material. However, in the present study, the compositional content of silica in enstatite was found to be around 60%, which might be retarded the deposition of HAp on the surface of the enstatite scaffold [43].
Forsterite contains more compositional magnesium content than enstatite and the former show higher apatite deposition ability than the latter, which may be due to the following reasons. The dissolution in forsterite leads to the breaking of Mg–O bonds ultimately causes the breakdown of the crystal structure and liberates Mg and Si ion. The breaking of the Mg–O bond occurs because of isolated silica tetrahedron bonded with magnesium consisting of octahedral coordination in forsterite. In contrast, the chain linkage of silica tetrahedron with two distinct magnesium sites which does not allow the complete elimination of ions from the crystal during Mg–O bond breaking in enstatite [44]. On account of the above statements, silica is not activated on the surface; thereby reduced apatite deposition took place on enstatite than forsterite.
It was found that the compressive strength associated with enstatite is in the range of trabecular bone, whereas forsterite appears in the range of cortical bone. Similarly, Young’s modulus of enstatite is equivalent to cancellous bone and forsterite lies in between spongy and compact bone. The observed results are found to lie in the range of existing results [11]. The possible reasons behind the mechanical strength are summarized as follows: firstly, enstatite was unable to induce hydroxyapatite mineralization, which can act as reinforcement and increase the density of the scaffold [45]. Second, hardness associated with forsterite is more than enstatite, which leads to slow degradation of forsterite compared to enstatite [46]. Even though both the materials follow orthorhombic crystal system, forsterite belongs to olivine group consist of isolated (SiO4)4− attached to the metal cation directly whereas enstatite belongs to pyroxene group possess the single chain of (SiO3)2− with edge corned octahedron placed between different cationic sites [47, 48]. Thus, it can be suggested that crystal structure also plays a vital role in predicting the mechanical stability of biomaterials.
The cell wall of both Gram-positive and Gram-negative test strains are negatively charged, which binds to the positively charged magnesium ions present in both the biomaterials and thus altering the cell permeability. Magnesium ions are reported to produce reactive oxygen species (ROS), which stimulates the peroxidation of lipids in bacteria [49]. This prevents the transport of essential ions into the cell [50], resulting in leakage of proteinaceous compounds and other intracellular constituents and causing the death of the microorganisms. It was observed with the previous findings that the antibacterial activity increases with the increase in the pH of the medium [51]. Rocchietta et al., reported that forsterite showed growth of microorganisms even at the concentration of 25 mg/mL [52]. In the present study, both magnesium silicates (forsterite and enstatite) showed good antimicrobial activity even at a very low concentration that is 2 mg/mL.
An ideal material developed for orthopaedic applications should maintain cell viability and support the propagation of the cells over time [53]. In addition, the appropriate cell phenotype to be loaded in the materials is the crucial step to support neo bone tissue formation at the bone defect region. In this perspective, bone marrow-derived mesenchymal stromal cells (BMSCs) population is the most suitable candidate as it is the precursor for the bone cells including osteoblast and osteocyte. Apart from that, this cell population was widely established for bone tissue engineering owing to its stable characteristics i.e., express unique mesenchymal stem cell surface markers such as CD44, CD105, CD90 and CD73, plastic adherent and trilineage differentiation potential (Osteogenic, chondrogenic and adipogenic) as well as maintaining its fibroblastic such as morphology [54]. These characteristics are strongly recommended by The International Society for Cellular Therapy prior to considering the application of mesenchymal stem cell in tissue engineering [55]. The enstatite and forsterite scaffolds seeded cells have shown a 1.7 and 1.5 fold increase in cell proliferation, respectively, at day 14 comparing to day 1. This significant increase (p < 0.01) on day 14 indicates that the scaffolds possess an optimal surface for the cells to attach and grow. This finding indicates the biocompatibility of magnesium silicates. This is because, the magnesium ion (Mg2+) is predominately involved in bone remodelling and skeletal tissue development. Furthermore, Mg2+ may enhance mesenchymal stem cell attachment, proliferation and differentiation into osteoblast as this phenomenon has been evidenced in our published article [54]. These findings are also comparable to materials made of calcium-magnesium-silicates. It was shown that the calcium ion (Ca2+) on the surface of the material can performance as ligand similar to Mg2+ where it favours adsorption of proteins associated with osteoblast via positive electrostatic attraction [56]. Therefore, the calcium-magnesium-silicates composite could exert a positive effect on cell attachment and spreading as similar to magnesium silicates.
Conclusion
In this work, forsterite and enstatite were successfully prepared through the combustion route using citric acid as a fuel. The methodology assisted in reducing the calcination temperature of both magnesium silicates. The SEM and TEM observations showed a rough surface with agglomerated particle morphology and high crystalline nature of enstatite and forsterite. Biomineralization studies indicated that enstatite was unable to induce apatite deposition over the immersed surface whereas forsterite showed the existence of phosphate and carbonate groups after four weeks of immersion in SBF. Forsterite was found to be more mechanically stable than enstatite due to differences in their degradation rate when immersed in SBF. Both the magnesium silicate ceramics exhibited distinct mechanical strength which was found to be equivalent to cortical bone (forsterite) and cancellous bone (enstatite) even after immersion in SBF for 28 days. The antibacterial studies revealed that magnesium silicates have the potential to inhibit the growth of bacteria. The cytocompatibility study revealed that the bioceramic scaffolds have the potential to stimulate the growth and proliferation of hBMSCs. Thus, the enstatite and forsterite scaffolds seeded with human bone marrow stromal cells have shown a 1.7 and 1.5-fold increase in cell proliferation after two weeks as compared to day 1. These findings confirm that both the silicate scaffolds have biocompatible nature. It is suggested that the biomineralization ability of magnesium silicates can be enhanced by fabricating their composites with calcium-containing biomaterials. The current report concludes that forsterite can be a promising biomaterial for load/stress bearing applications or as a coating over metallic and polymeric implants for the prevention of bacterial infections. Therefore, forsterite can be recommended for further analysis involving animal study before being translated to a clinical trial.
Materials and methods
Sol–gel combustion
Appropriate amounts of Mg(NO3)2.6H2O (98%, SDFCL), Si(OC2H5)4 (TEOS, 98%, Sigma Aldrich) and stoichiometric quantity (as per O/F ratio) of citric acid were dissolved separately in double distilled water to prepare stock solutions.
All the stock solutions were mixed at equal volume in a beaker and stirred constantly on a magnetic stirrer. A transparent layer of TEOS was noticed over the surface of the solution. To achieve a homogeneous mixture, the pH of the solution was adjusted to 1.7. This resulted in the acceleration of hydrolysis reaction followed by polycondensation leading to the formation of a gel-like structure near the corners of the beaker. Finally, the reaction mixture was stirred for 30 min at 70 °C followed by continuous stirring at room temperature. The appearance of highly viscous gel in the beaker indicated the completion of the sol–gel process which was aged at room temperature and then dried at 120 °C. The dried gel was decomposed in a preheated muffle furnace at 400 °C. The combusted precursor was calcined at different temperatures to study the phase evolution of forsterite and enstatite.
Characterization
A thermal study of the as-prepared gel powder was carried out with the help of Universal V4.5A TA instrument (SDT Q600 V20.9 Build 20). The different functional groups of the silicate ceramics were determined through Shimadzu (IR Affinity 1) Fourier Transform Infrared Spectrometer. The evolution of phase purity of the magnesium silicate ceramics was analyzed by using Brucker D8 Advanced Powder X-ray diffractometer with Copper Kα radiation filter at the wavelength of 1.5406 Å. Surface topography/elemental analysis was analyzed with the help of Scanning Electron Microscope/Energy Dispersive X-ray spectroscopy of Hitachi S-3400 N model at an accelerating voltage of 20 kV. The particle size/selected area electron diffraction was investigated by utilizing 200 kV FEI-Tecnai G2 20 S-TWIN High-Resolution Transmission Electron Microscope. The evaluation of compressive strength and young's modulus were done using the universal testing machine (UTM) INSTRON 8801 as per ASTM standard procedure.
In-vitro studies
The biomineralization study was carried out in simulated body fluid (SBF). The preparation of SBF was done by using Tris(hydroxymethyl) aminomethane (99.8%, SDFCL), Sodium sulphate anhydrous (99.5%, SDFCL), Sodium Chloride (99%, SDFCL), Sodium bicarbonate (99%, Nice), Potassium chloride (99.5%, SDFCL), Magnesium chloride (99%, SDFCL), Di-potassium hydrogen orthophosphate (99%, SDFCL), Concentrated hydrochloric acid (SDFCL) and Calcium chloride (98%, QFC) as per Kokubo et al., procedure [57]. Enstatite and forsterite powders were uniaxially pressed at a pressure of 5 ton to prepare the scaffolds as per ASTM standard (ASTM F2883-11) (2:1 diameter to height ratio), with 12 mm diameter and 6 mm height. The bioceramic scaffolds were then annealed in a furnace at 900 °C for 3 h (after heating up at a rate of 5 °C/min from room temperature). The scaffolds were immersed in the SBF with a pH of 7.4 and incubated at 37 °C to carry out bioactivity, mechanical and resorbability studies. The immersed cylindrical scaffolds were dried at 150 °C for a day after each interval time (7, 14, 21 and 28 days) and characterized by XRD for examining apatite deposition over their surface. Further, the scaffolds were weighed to estimate resorbability with simultaneous degradation of material and deposition of HAp through the weight loss-gain parameter.
After bioactivity and resorbability studies, the scaffolds were subjected to mechanical strength evaluation using a universal testing machine (INSTRON) at a compression rate of 1 mm/min and an extension rate of 0.6 mm.1.5 g of pure enstatite and forsterite materials were made as scaffolds with the ratio of 2:1 diameter to height using hydraulic pellet press at 15,000 Ib/in2 as per the ASTM standard procedure. These studies were carried out in triplicates for each sample and the results are demonstrated as a mean with standard deviation.
Antibacterial activity assay
The ability of forsterite (Mg2SiO4) and enstatite (MgSiO3) to inhibit the growth of a wide spectrum of clinical pathogens was investigated. All the target strains were revitalized on nutrient agar medium and incubated for 24 h. Freshly grown cultures were inoculated in nutrient broth for 24 h at 37 °C. Bacterial culture suspension was diluted with sterile physiological solution (0.8% saline solution) to 108 CFU/mL (turbidity = McFarland barium sulphate standard 0.5). The culture density was adjusted to an optimum level of 1.00 O.D. for broth dilution assay.
The broth dilution technique was adopted to evaluate the antimicrobial activity of both the magnesium silicate biomaterials due to their insoluble behaviour. 100 mL of Luria Bertania (LB) broth was prepared in 250 mL Erlenmeyer flask containing a varying concentration of enstatite and forsterite (0.5, 1and 2 mg/mL) along with 500 µL of clinical pathogens in each flask. Control flasks containing LB broth and test strains were also prepared. All the flasks were incubated in a rotary shaker at 180 rpm for 24 h at 37 °C [11]. Post incubation the bacterial growth, optical density (OD) was measured at an absorbance of 600 nm using ELISA reader (Biotek-elx800) and percentage of inhibition was calculated using the formula:
CF—Control broth containing test strain.
BCF—Final concentration (2000 µL) of biomaterial containing an organism.
To analyze the relationship between the concentration of biomaterial and bacterial growth inhibition, different dilutions were spread plated on the Mueller Hinton agar plate. The interaction between the clinical pathogens and magnesium silicate biomaterials was studied using a scanning electron microscope (Zeiss Evo 18) and then the samples were viewed under SEM at 10 kV.
Cell culture
hBMSCs isolation and culture
Ethical approval was granted by the University of Malaya Medical Centre (UMMC) ethics committee (Ethics No: 20164-2398) to obtain patient bone marrow samples. Human bone marrow aspirates were obtained from subjects (50–70 years old) undergoing total knee/hip replacement. Informed consent was obtained before acquiring samples. The human bone marrow-derived mesenchymal stromal cells (hBMSCs) isolation methods were adopted from a published article [54]. Briefly, bone marrow mononucleated cells (MNCs) were purified using standard Ficoll-Pague gradient centrifugation (density 1.073 g/mL) according to the manufacturer’s instruction (GE Healthcare Bio-Sciences, USA). The density gradient centrifugation was performed at 2200 rpm for 25 min. The middle layer containing MNCs was isolated and washed three times with PBS (1X) (Gibco, Invitrogen, USA). The MNCs were then suspended in culture medium DMEM-LG (Gibco) containing 100 U/mL of penicillin and 100 µg/mL of streptomycin supplemented with FBS (Gibco). Cell number and viability were enumerated using the Trypan blue exclusion method. About 1 X 106 cells were seeded onto T-75 culture flask and incubated at 37 ºC in 5% CO2 with 95% humidity. For subsequent passaging, the cells in passage-0 (P0) were washed with PBS (1X) and then incubated in trypsin (Gibco) for 3 min in a CO2 incubator at 37 °C until complete cell detachment observed. Harvested P0 cells were sub-cultured into passage-1 (P1) and the culture medium was changed every 3 days interval.
Cell seeding
For sterilization, enstatite and forsterite scaffolds were sent for 25 kGy gamma irradiation (Nuclear Agency of Malaysia). The cell seeding was performed using a published protocol [54]. Briefly, hBMSCs were enzymatically detached using 3 mL of trypsin after reaching 80% of confluence at P1. A cell suspension was prepared and seeded onto the enstatite and forsterite scaffolds in low attachment twelve-well plate in a dropwise manner at the seeding density of 1 X 106 cells/mL. The cell-seeded enstatite and forsterite scaffolds were cultured using culture medium DMEM-LG (Gibco) containing 100 U/mL of penicillin and 100 µg/mL of streptomycin supplemented with FBS (Gibco).
Cell attachment
Scanning electron microscopy analysis
Scanning electron microscopy (SEM) analysis was performed to observe surface topography of hBMSCs (n = 3) seeded on enstatite and forsterite scaffolds using a published protocol [53]. The samples at day 14 were fixed overnight in 4% glutaraldehyde in 0.1 M cacodylate buffer and post-fixed for 1 h in 1% aqueous osmium tetroxide. These samples were washed with three consecutive steps in distilled water before being dehydrated through a graded ethanol series (50, 70, 80, 90, 95 and 100%). The samples were subsequently dried at a critical point using critical point drier (Bal Tec, CPD030). The samples were mounted on an aluminium stub and sputter-coated with gold before being examined using a digital scanning electron microscope (JSM 6400; JEOL, Tokyo, Japan).
Cell proliferation analysis
Cell proliferation in enstatite and forsterite scaffolds (n = 6) was assessed using the colorimetric indicator Alamar Blue (AB) cell proliferation/viability assay (Gibco). The assay was performed based on the AB reduction on days 1, 3, 7 and 14. AB was directly added into the media in all preparation at a final concentration of 10% and incubated for 12 h. After incubation, 100 μL of the medium from each sample was transferred into a 96-well plate in duplicates. AB added to hBMSCs monolayer was served as a baseline control and samples without AB served as a blank. Absorbance in each well was measured at 570 and 600 nm (reference wavelength) using a microplate reader (Epoch, USA). The corrected absorbance readings were calculated by subtracting the individual reference wavelength from the respective measured wavelength and presented as mean ± 2 standard deviations (SD).
References
J. Gargiuli, A. Gill, G. Lillie, M. Schoenleber, J. Pearson, G. Kyriakou, P. Vadgama, Surfaces and interfaces for biomaterials, (Woodhead publishing, 2005), pp. 103–149.
M. Diba, O.M. Goudouri, F. Tapia, A.R. Boccaccini, Curr. Opin. Solid State Mater. Sci. (2014). https://doi.org/10.1016/j.cossms.2014.02.004
M. Nabiyouni, T. Brückner, H. Zhou, U. Gbureck, S.B. Bhaduri, Acta Biomater. (2018). https://doi.org/10.1016/j.actbio.2017.11.033
E.M. Bueno, J. Glowacki, Nat Rev Rheumatol. (2009). https://doi.org/10.1038/nrrheum.2009.228
S. Ni, J. Chang, J. Biomater. Appl. (2009). https://doi.org/10.1038/nrrheum.2009.228
D. Goeuriot, J.C. Dubois, D. Merle, F. Thevenot, P. Exbrayat, J. Eur. Ceramic Soc. (1998). https://doi.org/10.1016/S0955-2219(98)00117-4
M. Vallet Regi, A.J. Salinas, J. Roman, M. Gil, J. Mater. Chem. (1999) https://doi.org/10.1039/A808679F
M.A. Naghiu, M. Gorea, F. Kristaly, M. Tomoaia-Cotisel, Ceram. Silik. 58, 303 (2014)
M.A. Naghiu, M. Gorea, E. Mutch, F. Kristaly, M. Tomoaia-Cotisel, J. Mater. Sci. Technol. (2013). https://doi.org/10.1016/j.jmst.2013.04.007
R. Choudhary, P. Manohar, J. Vecstaudza, M.J. Yáñez-Gascón, H.P. Sánchez, R. Nachimuthu, J. Locs, S. Swamiappan, Mater. Sci. Eng. C (2017). https://doi.org/10.1016/j.msec.2017.03.308
R. Choudhary, A. Chatterjee, S.K. Venkatraman, S. Koppala, J. Abraham, S. Swamiappan, Bioact. Mater. (2018). https://doi.org/10.1002/jbm.a.36925
R. Choudhary, S.K. Venkatraman, I. Bulygina, F. Senatov, S. Kaloshkin, N. Anisimova, M. Kiselevskiy, M. Knyazeva, D. Kukui, F. Walther, S. Swamiappan, Mater. Sci. Eng. C (2021). https://doi.org/10.1016/j.msec.2020.111456
M. Gorea, M.A. Naghiu, A. Avram, I. Petean, A. Mocanu, M. Tomoaia-Cotisel, Rev. Chim. (2020) https://doi.org/10.37358/RC.20.2.7935
M. Kharaziha, M.H. Fathi, J. Mech. Behav. Biomed. Mater. (2010). https://doi.org/10.1016/j.jmbbm.2010.06.003
R. Lakshmi, R. Choudhary, D. Ponnamma, K.K. Sadasivuni, S. Swamiappan, Bull. Mater. Sci. (2019). https://doi.org/10.1007/s12034-019-1814-4
R. Choudhary, S.K. Venkatraman, I. Bulygina, A. Chatterjee, J. Abraham, F. Senatov, S. Kaloshkin, A. Ilyasov, M. Abakumov, M. Knyazeva, D. Kukui, F. Walther, S. Swamiappan, J. Asian Ceram. Soc. (2020). https://doi.org/10.1080/21870764.2020.1807695
S.K. Venkatraman, S. Swamiappan, Chem. Select (2019). https://doi.org/10.1002/slct.201902780
Z. Wu, K. Zheng, J. Zhang, T. Tang, H. Guo, A.R. Boccaccini, J. Wei, J. Mater. Chem B. (2016). https://doi.org/10.1039/C6TB02429G
X. Jin, J. Chang, W. Zhai, K. Lin, J. Am. Ceram. Soc. (2011). https://doi.org/10.1111/j.1551-2916.2010.04032.x
S. Yamamoto, T. Nonami, H. Hase, N. Kawamura, J. Aust. Ceram. Soc. 48, 180 (2012)
A. Saberi, Z. Negahdari, B. Alinejad, F. Golestani-Fard, Ceram. Int. 35, 1705 (2009)
H.B. Bafrooei, T. Ebadzadeh, H. Majidian, Ceram. Int. (2014). https://doi.org/10.1016/j.ceramint.2013.10.025
S. Ni, L. Chou, J. Chang, Ceram. Int. (2007). https://doi.org/10.1016/j.ceramint.2005.07.021
F. Tavangarian, R. Emadi, Mater. Res. Bull. (2010). https://doi.org/10.1016/j.materresbull.2009.12.032
R.Y.S. Zampiva, L.H. Acauan, L.M. Dos Santos, R.H.R. De Castro, A.K. Alves, C.P. Bergmann, Ceram. Int. (2017). https://doi.org/10.1016/j.ceramint.2017.08.201
S. Ramesh, A. Yaghoubi, K.S. Lee, K.C. Chin, J. Purbolaksono, M. Hamdi, M.A. Hassan, J. Mech. Behav. Biomed. Mater. (2013). https://doi.org/10.1016/j.jmbbm.2013.05.008
P. Ptacek, K. Lang, F. Soukal, T. Opravil, E. Bartonickova, L. Tvrdik, J. Eur. Ceram. Soc. (2014). https://doi.org/10.1016/j.jeurceramsoc.2013.08.007
A. Goel, D.U. Tulyaganov, E.R. Shaaban, C.S. Knee, S. Eriksson, J.M. Ferreira, Ceram. Int. 35, 1529 (2009)
T. Van Long, Ceram. Int. (2008). https://doi.org/10.1016/j.ceramint.2007.06.014
L. Lin, Y. Min, S. Chaoshu, Z. Weiping, Y. Baogui, J. Rare. Earth. 24, 104 (2006)
F. Alam, K. Balani, J. Mech. Behav. Biomed. Mater. (2017). https://doi.org/10.1016/j.jmbbm.2016.10.009
S.P. Sawan, G. Manivannan, Antimicrobial/Anti-Infective Materials: Principles and Applications, (CRC Press, Florida), pp. 346.
E. Novitskaya, J.P. Kelly, S.B. Bhaduri, O.A. Graeve, Int. Mater. Rev. (2021). https://doi.org/10.1080/09506608.2020.1765603
A. Trampuz, D.R. Osmon, A.D. Hanssen, J.M. Steckelberg, R. Patel, Clin. Orthop. Relat. Res. (2003). https://doi.org/10.1097/01.blo.0000087324.60612.93
A. Varma, A.S. Mukasyan, A.S. Rogachev, K.V. Manukyan, Chem. Rev. (2016). https://doi.org/10.1021/acs.chemrev.6b00279
R.D. Purohit, A.K. Tyagi, J. Mater. Chem. (2002). https://doi.org/10.1039/B103461H
R.B. Rao, H. Singh, Def. Sci. J. 46, 327 (1996)
S. Sasikumar, R. Vijayaraghavan, J. Mater. Sci. Technol. (2010). https://doi.org/10.1016/S1005-0302(11)60010-8
E. Landi, A. Tampieri, M. Mattioli-Belmonte, G. Celotti, M. Sandri, A. Gigante, G. Biagini, J. Eur. Ceram. Soc. 26, 2593 (2006)
X. Chen, J. Ou, Y. Wei, Z. Huang, Y. Kang, G. Yin, J. Mater. Sci: Mater Med. (2010). https://doi.org/10.1007/s10856-010-4025-5
M. Cerruti, N. Sahai, Rev. Mineral Geochem. (2006). https://doi.org/10.2138/rmg.2006.64.9
G. Krishnamurithy, M.R. Murali, M. Hamdi, A.A. Abbas, H.B. Raghavendran, T. Kamarul, Regen. Med. (2015). https://doi.org/10.2217/rme.15.27
S.M. Best, A.E. Porter, E.S. Thian, J. Huang, J. Eur. Ceram. Soc. (2008). https://doi.org/10.1016/j.jeurceramsoc.2007.12.001
E.H. Oelkers, in Growth, Dissolution and Pattern Formation in Geosystems, ed. by B. Jamtiveit, P. Meakin, (Springer, Dordrecht), p. 253
T. Wang, Z. Feng, Mater. Lett. (2005). https://doi.org/10.1016/j.matlet.2004.08.048
K.S. Lee, K.C. Chin, S. Ramesh, J. Purbolaksonoa, M.A. Hassan, M. Hamdi, W.D. Teng, J. Ceram. Process. Res. 14, 131 (2013)
F. Liebau, Structural chemistry of silicates: structure, bonding, and classification, (Springer Science & Business Media, Berlin, 2012), p. 278
L.M. Anovitz, A.J. Rondinone, L. Sochalski-Kolbus, J. Rosenqvist, M.C. Cheshire, J. Colloid. Interf. Sci. (2017). https://doi.org/10.1016/j.jcis.2017.01.052
Q. Li, S. Mahendra, D.Y. Lyon, L. Brunet, M.V. Liga, D. Li, P.J. Alvarez, Water Res. (2008). https://doi.org/10.1016/j.watres.2008.08.015
I.S. Kwun, Y.E. Cho, R.A.R. Lomeda, H.I. Shin, J.Y. Choi, Y.H. Kang, J.H. Beattie, Bone (2010). https://doi.org/10.1016/j.bone.2009.11.003
D. Raafat, H.G. Sahl, Microb. Biotechnol. (2009). https://doi.org/10.1111/j.1751-7915.2008.00080.x
I. Rocchietta, M. Simion, M. Hoffmann, D. Trisciuoglio, M. Benigni, C. Dahlin, Clin. Implant. Dent. Relat. Res. (2016). https://doi.org/10.1111/cid.12267
S. Dhivya, A. Keshav Narayan, R. Logith Kumar, S. Viji Chandran, M. Vairamani, N. Selvamurugan, Cell. Proliferat. (2018) https://doi.org/10.1111/cpr.12408
G. Krishnamurithy, S. Mohan, N.A. Mansor, M.R. Murali, H.R.B. Raghavendran, R. Choudhary, S. Swamiappan, T. Kamarul, PLoS One (2019). https://doi.org/10.1371/journal.pone.0214212
D.F. Stroncek, P. Jin, D.H. McKenna, M. Takanashi, M.J. Fontaine, S. Pati, R. Schafer, E. Peterson, E. Benedetti, J.A. Reems, Front. Cell Dev. Biol. (2020). https://doi.org/10.3389/fcell.2020.00458
X. Zhang, C. Zhang, W. Xu, B. Zhong, F. Lin, J. Zhang, Q. Wang, J. Ji, J. Wei, Y. Zhang, Int. J. Nanomed. (2015). https://doi.org/10.2147/IJN.S92598
T. Kokubo, H. Takadama, Biomaterials (2006). https://doi.org/10.1016/j.biomaterials.2006.01.017
Acknowledgments
The authors acknowledge the Vellore Institute of Technology (VIT) management for the support and CAMPT-VIT for helping with the mechanical studies. The authors also thank DST-FIST for the XRD and SEM-EDX facility. The authors gratefully acknowledge the financial support from the European Union’s Horizon 2020 research and innovation program under the grant agreement No. 857287.The authors would like to express their highest gratitude to Ministry of Higher Education for fundamental research grant scheme (FRGS)—FRGS/1/2016/SKK08/UM/02/20.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Venkatraman, S.K., Choudhary, R., Vijayakumar, N. et al. Investigation on bioactivity, mechanical stability, bactericidal activity and in-vitro biocompatibility of magnesium silicates for bone tissue engineering applications. Journal of Materials Research 37, 608–621 (2022). https://doi.org/10.1557/s43578-021-00450-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43578-021-00450-9