
Abstract
Liquid phase electron microscopy is a new analytical method that has opened up a rapidly emerging field of research during the past decade. This article discusses this new microscopy modality within the context of imaging eukaryotic cells, bacteria, proteins, viruses, and biomineralization processes. The obtained resolution is typically not a function of the instrument, rather it is limited by the available electron dose within the limit of radiation damage. Therefore, different types of samples are best imaged with different electron microscopy (EM) modalities. The obtained information differs from that acquired with conventional EM as well as cryo-electron microscopy. This article gives an overview of achievements thus far in this area and the unique information that has been obtained. A discussion on potential future developments in the field, and technological advancements required to reach those goals conclude the article.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Conventional electron microscopy requires biological samples to be in a fully solid state to withstand the high vacuum in the chamber of the electron microscope, which is usually achieved by either fixation, dehydration, and embedding or rapid freezing of hydrated specimens. However, life happens in water. As such, samples would intuitively be studied in their native liquid environment with electron microscopy, as is done with light microscopy. The desire to image biological specimens in their natural, liquid state was already expressed at the onset of electron microscopy in the 1940s, but liquid phase electron microscopy (LP-EM) with nanometer resolution has become available to the broad electron microscopy community only in the recent decade.1,2 Since then, the research community has observed an increase in publications on different biological samples imaged in liquid with transmission electron microscopy (TEM), and scanning TEM (STEM), in addition to several innovations in scanning electron microscopy (SEM).1,3–6
Here, a brief overview of current possibilities in LP-EM of biological samples is presented. Due to the constraints of this article, it is impossible to provide a comprehensive overview, so only highlights are given. We will discuss potential future developments of the field, and aim to answer the questions regarding the benefits and unique information that may be obtained from LP-EM.
LP-EM technology
Two different technological principles for LP-EM now exist since the early days of electron microscopy:7,8 open environment systems maintaining a liquid layer directly in the specimen chamber of the electron microscope, and closed systems in which the liquid is separated from the vacuum by at least one thin membrane through which the electron beam propagates. Variable pressure, also known as environmental SEM, has been available to the broad community the longest,9,10 but could only achieve limited resolutions for samples in liquid. This was finally improved in the mid 2000s by using STEM detection11 providing nanometer resolution on materials embedded in a thin liquid layer due to efficient detection in transmission mode.12 A liquid capsule with a polymer membrane was used in SEM in the early 2000s, though this method also had limited resolution.13 The ability to image samples fully embedded in liquid with nanometer resolution became available in late 2000s,14 using closed systems with silicon nitride membranes.15 The achievable resolution of LP-EM for nanoparticle-labeled proteins in a whole eukaryotic cell using a liquid flow specimen holder2 reached ∼4 nm, a similar resolution to that achieved for imaging labeled bacteria in a static liquid enclosure (no flow).16
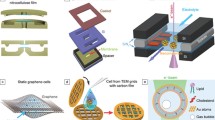
In 2010, a third principle was introduced, atmospheric SEM (ASEM). ASEM consists of a modified cell culture dish, which can be placed in an atmospheric environment, and then probed by an electron beam through a single SiN window. Examination with light microscopy can be done in parallel.17 In the following years, the most important step forward was the replacement of SiN membranes with much thinner, single- or multilayer graphene sheets to either cover or fully enclose the sample18–20 (see Figure 1); graphene also had the ability to reduce radiation damage by quenching excited states that would otherwise result in damaging chemical radicals.21,22 Three-dimensional (3D) TEM has been demonstrated either via specimen tilting23 or using single-particle techniques,24 while a 360° tilting stage was integrated in SEM.25 Time-resolved LP-EM is now routinely used in materials science and chemistry,3 but radiation damage still poses a challenge. TEM offers the ability for high-speed microscopy, since modern TEM cameras achieve millisecond frame rates. Time-resolved STEM is also possible at video frequency and beyond.26

Scheme of liquid phase electron microscopy (LP-EM) of four different classes of biological samples. Biological objects for LP-EM cover a wide range of thicknesses, ranging from 5 to 100 nm for proteins and small viruses, 0.1 to 2 μm for most bacteria, to thicker than 2 μm for eukaryotic cells and layers of tissues, including tissues with biominerals such as bone. To protect the samples against the vacuum inside the specimen chamber, samples are enclosed between microchips covered with SiN membranes, or “wrapped” with graphene sheets mechanically stabilized with transmission electron microscopy (TEM) grids or with a SiN microchip. Imaging a sample between two microchips requires a specialized flow holder, whereas samples in a graphene liquid cell can be placed in any TEM holder. Alternatively, the samples can be placed or immobilized on a supporting SiN microchip and examined in wet state in an “open” EM system—this can be an environmental or atmospheric scanning electron microscope (ESEM or ASEM). Prior to EM, it is possible and advisable to record light microscope images.
Sample considerations
Due to the radiation sensitivity of biological materials combined with the electrochemical beam effects induced by LP-EM,27 the capabilities of LP-EM critically depend on the dose tolerance of the specimen.5 For non-fixed biological materials, the cryo-TEM electron dose limit of 10 e– Å–2 is often used,28 while chemically fixed structures can tolerate doses29 over 103 e– Å–2. However, detailed knowledge about the dose tolerance in liquid is still lacking. Smaller sample features tend to have a lower dose tolerance,30 while a higher dose tolerance was demonstrated in liquid than in ice.31 The degree of radiation damage depends on the electron flux, since reversible effects, such as charge creation, can be equilibrated at low flux.22,32,33
LP-EM experiments need to be precisely tuned to achieve nanoscale resolution within the electron dose limit,5,34 which determines the number of available electrons required to achieve a sufficient signal-to-noise level. The electron dose D scales with the resolution δ as δ ∝ D1/4.34 This relation implies that the dose levels rapidly reduce with even a mild relaxation of the resolution requirement. Since the signal-to-noise level decreases with increasing sample thickness, it is logical to classify biological samples in a hydrated state by their thickness range (see Figure 1). The optimal experimental settings are different for each class of thickness.
Nanometers- to hundreds-of-nanometers-thick specimens include proteins, large protein complexes, elongated DNA strands, and most virus particles. The carbon-based structures of these samples are usually viewed with TEM. To achieve the best possible resolution, it is critical to minimize the thickness of the liquid layers as much as possible and use the thinnest possible material for the enclosure, preferably graphene or another ultrathin material. The spatial resolution amounts to ∼4 nm for a low electron dose at 1 e– Å–2 for contrast on carbon in a thin water layer at 200 keV beam energy, using atomically thin liquid enclosing membranes, such as those constructed of graphene.4 Applying diffraction techniques for protein crystals in liquid and subnanometer resolution is also of interest and has already been demonstrated in the 1970s.30
Submicrometer-thick specimens include large viruses, bacterial cells, large organelles, thin protrusions of eukaryotic cells, and ultrathin tissue sections. In practice, TEM is mostly used, although it may not always be optimal when the sample is thicker than half of the mean-free-path-length of elastic electron scattering (160 nm at 200 keV beam energy).34 For this sample class, the window thickness is also critical. The resolution can be improved by energy filtering, albeit at increased electron dose,35 or ideally by chromatic aberration correction.36 Alternatively, thicker carbon-based samples can be imaged with bright-field STEM.37,38
Micrometers-thick specimens include whole eukaryotic cells and tissue sections. These are best studied by using specific labels consisting of materials of high atomic number (Z) with STEM2 because the resolution for a carbon-based material is several tens of nanometers.39 Enclosing windows of 50-nm-thick silicon nitride do not add much to the total thickness, and are thus not critical. A particularly elegant solution is to enclose samples, grown or bound, to a SiN membrane under a sheet of graphene,18 closely wrapping the sample such that maximal resolution is achieved on thin cellular areas, despite their much thicker central areas.40
Biominerals form the fourth class of specimens. This class, which includes bone, has different material composition than cells and proteins and includes a major fraction of elements with a higher Z than carbon and water. The thickness argument is thus different than for the carbon- and water-based materials in the former three sample classes.
Class 1 samples: Proteins, DNA, and virus particles
The literature of the past decade contains numerous examples showing LP-EM of small biological objects, such as single proteins,41,42 so-called acrosomal bundles of cross-linked actin filaments31 (Figure 2a–b), microtubules22 (Figure 2c), actin filaments,31 ferritins,42 viral assemblies,43 and antibodies.44 The main purpose of most of those studies seems to have been exploring the capabilities of LP-EM, rather than obtaining unique biological information. Improved structural information for these samples can be obtained via conventional- or cryo-EM, although LP-EM has shown a much better dose tolerance31 under specific conditions equilibrating reversible radiation processes.22,32

Examples of liquid phase electron microscopy (LP-EM) of thin biological specimens of type 1. (a) Overview transmission elecron microscope image of agglomerated 80-nm diameter acrosomal bundles in liquid. (b) An acrosomal bundle showing the repetitive units. (c) A microtubule in a graphene liquid cells with clearly visible protofilaments. (d) Time-resolved LP-EM images (bottom) showing a DNA zipper-up hybridization process, starting with a contact of bases located near the middle of the strands. The arrows indicate rotation. The schematic diagrams above the LP-EM images serve as a guide to the eye showing time-dependent conformations in the top row and base-pair alignment in the middle row.47 (a, b) Reprinted with permission from Reference 31. © 2012 Elsevier. (c) Adapted with permission from Reference 22. © 2018 American Chemical Society. (d) Reprinted with permission from Reference 47. © 2020 National Academy of Sciences.
The key advantage of LP-EM for this type of sample is the prospect of time-resolved studies to examine the role of structural dynamics in protein function. Due to radiation damage, it is highly challenging to study biological processes under physiological conditions. Despite these challenges, several groups have been able to study dynamics, such as the movement of the heads of myosin motor proteins,8 the nucleation and growth of lysozyme crystals,45 virus mobility,46 and the process of DNA changing its conformation from single strands to secondary structure47 (Figure 2d). New insights can be expected from this type of experiment for principles behind conformational changes in biological macromolecules.
Class 2 samples: Bacterial cells
Liquid enclosures with silicon nitride windows can be used to image bacteria.48 Graphene liquid cells were first demonstrated with enclosed bacteria.49 A particularly elegant example was the visualization of magnetic fields in magnetotactic bacteria using off axis holography in TEM.50 The interaction of a bacteriophage and its host bacterium has been studied using 3D viewing in LP-EM.23 Conducting live-cell LP-EM experiments using bacteria would be the next important goal. Since bacteria have a robust cell wall, the outer cell wall could potentially be irradiated, while carefully preventing the cell interior from radiation.51 Note that the claim of live cell imaging needs to be supported by the key criteria of life, namely, the ability to reproduce.52 However, a “blind” examination of the survival rate in a bulk bacterial colony is insufficient. Instead, bacteria that have actually been irradiated need to be examined exclusively. The typical radiation level in TEM is four orders of magnitude above the lethal dose for bacteria,53 so it is unlikely that true time-resolved, live cell LP-EM will work unless special measures are taken to reduce the dose and mitigate damage effects. Alternatively, biochemical reactions could be studied at lethal dose in a limited time window.
Class 3 samples: Eukaryotic cells
LP-EM provides the ability to study cells in an “as-native-as-possible” state. This implies that cells are kept intact, and thus represent relatively thick samples compared to the usual thin sections prepared for biological TEM. To exploit the high resolution of EM in micrometers-thick cellular samples, most LP-EM studies use electron-dense labels, for instance, gold nanoparticles or fluorescent quantum dots.54 These are well-suited for correlative light microscopy and LP-EM,55 and specifically for binding to targeted structures of the eukaryotic cells. The information differs from conventional TEM, which is optimized for studying ultrastructure, but highlights specifically labeled proteins, similar to the fluorescent labeling used for modern light microscopy. Since these nanoparticle labels can be detected with up to a few nanometers precision, the analysis of specific features, such as their spatial positions, density, or clustered arrangements, have provided new insights for different intracellular structures and functions, including the cytoskeleton, the endoplasmic reticulum, endocytosis mechanisms, or proteins involved in Ca2+ signaling.56,57 Of particular interest is the study of membrane proteins, which represent one of the most abundant and functionally varied classes of proteins accounting for ∼60% of today’s drug targets, directly in the intact plasma membrane. So far, two receptors for growth factors and two ion channels have been successfully examined, revealing functionally relevant grouping/patterning features,40,58 their stoichiometry and activation status12,59,60 (see example in Figure 3), and the effects of modern targeting drugs applied in cancer therapies.61 Furthermore, tissue samples have been investigated with LP-EM, including exocrine glands,62 dissociated single tumor cells from patient biopsies,58 and various mouse tissues.63

Examples of liquid phase electron microscopy (LP-EM) images of eukaryotic cells (sample type 2). Correlative light- and LP-EM of hydrated, intact breast cancer cells with quantum dot (QD) labeled HER2, an oncoprotein responsible for uncontrolled cell growth. (a) Direct interference contrast image of cells, most showing elongated protrusions of the plasma membrane, so-called membrane ruffles (see arrows). (b) Red QD-fluorescence high HER2 expressing cell (cell 1) and green fluorescence signals, identifying cancer stem cells (cell 2). (c) Environmental scanning electron microscope-scanning transmission electron microscope (ESEM-STEM) overview image recorded of the same cells. (d) High-resolution ESEM-STEM image (50,000× magnification) recorded at the position of the rectangle in (c), displaying the individual HER2-bound QDs (highlighted in yellow), preferentially accumulating on the membrane ruffles. Reprinted with permission from Reference 61. © 2017 American Society for Cell Biology.
Class 4 samples: Biomineralization
Another application of LP-EM is the study of biomineralization processes. Calcium carbonate nucleation has been monitored in the absence64 and presence of additives, both organic65 and inorganic.66 A range of simultaneous nucleation pathways were observed, including direct nucleation from solution and indirect transformation of amorphous and crystalline precursors.64 Direct transformation of amorphous calcium carbonate (ACC) to vaterite or aragonite was recorded for the first time; the secondary nucleus appeared at the surface of the ACC particle and grew in contact and at the expense of the ACC particle (Figure 4a–d). Nevertheless, in the presence of a high concentration of Mg2+, ACC transformed to calcite through a direct shape-preserving pathway.66 Calcite retained the shape of the amorphous phase, providing evidence for a mechanism of morphological control of crystallization that may be key in biomimetics.

Examples of biomineral samples studied with liquid phase electron microscopy (LP-EM), type 4 samples. (a) Amorphous calcium carbonate (ACC) particle. (b) Particle with secondarily nucleated vaterite plates forming on or in the amorphous particle. These plates grow at the expense of the ACC (c, d). Diffraction identifies the nucleated plates as vaterite [inset to (d)] Scale bars = 500 nm. (e) Atmospheric scanning electron microscope image of freshly sliced trabecular bone. (f) Immuno-gold labeled cathepsin K positive cell imaged at the location of the rectangle in (e). (a–d) Reprinted with permission from Reference 64. © AAAS. (e, f) Reprinted with permission from Reference 69. © 2019 Nature Publishing Group.
LP-EM emerged as a promising technique for studying the dynamics of bone mineralization. Aggregation of calcium phosphate prenucleation clusters, previously characterized by cryo-EM,67 have been recently monitored in liquid.68 The formation of small particles was visualized after 4 min of beam-exposure (average radii 10 nm) with movement and aggregate forming branched particle assemblies after 9 min.
A major challenge in the field is bridging the gap between the physiochemistry of the mineral precursors and the mineralization process at the cellular and tissue level. The small size and shape of the TEM specimen holder for LP-EM limits the culturable cell types (i.e., osteoclasts 150–200 μm in diameter), and therefore the visualization of certain processes. The ASEM presents an alternative for the study of larger samples, such as cell cultures and tissues, with minimal preparation. Samples were grown in disposable dishes with a SiN film window, lightly aldehyde-fixed, and kept in their liquid environment during the measurement. Using this technique, the onset of mineralization in primary culture osteoblasts was recently observed for the first time in liquid.69 Calcium phosphate patches (confirmed by correlative energy-dispersive spectroscopy (EDS) analysis) appeared after eight days from induction of differentiation. Moreover, direct observation of freshly sliced trabecular bone was possible prior to fixation, combining the use of in-solution immunolabeling methods56 to determine osteoclast activity69 (Figure 4e–f).
Outlook
The ability to image membrane proteins with a resolution below their own dimensions is still restricted to EM, even though super resolution fluorescence techniques approach the nanometer resolution range.70 It remains an empirical trial-and-error process to efficiently stabilize membrane proteins outside their environment; therefore, keeping them in the plasma membrane during imaging is the safest way to obtain undisturbed information from their native state. Other techniques exist for examining membrane proteins in whole cells, such as cryo-EM,71 but this technique is mainly used for studying the structure of proteins extracted from cells or in thin sections, and not for dynamic processes due to practical limitations. Future developments of LP-EM must include new strategies for versatile labeling systems in order to open the LP-EM “viewing window” for many more of the ∼5000 mammalian integral membrane proteins.
These labeling approaches should follow the established principle of both small proteins binding to the target and monodisperse, electron-dense nanoparticles. An enlarged repertoire of labels is needed, and could be created by using new combinations of specific, small molecules and peptides, and fitting peptide-tag modified nanoparticles.72 Besides the presently used ligands, small antibody mimetic compounds such as Affibodies, and fragments of antibodies could be applied in the future, in addition to others like designed ankyrin repeat proteins (DARPins), aptamers (oligonucleotides or peptides), or drugs. Such a label “toolbox” would allow for multiplexed labeling, and thus for the in situ study of protein interactions needed to improve our knowledge of membrane proteins, and support the development of personalized disease diagnosis and treatment.
Protein structure–function relationships involve highly complex interactions that require examination with a multitude of techniques, each adding a different “viewing angle.” LP-EM already provides unique information about biological structures in liquid state, and might contribute key information about conformational variability and its dynamics, a future aspect of protein science. Proteins tend to be viewed as distinct structural units, as suggested by protein crystallographic information. The real-life picture, however, is much more complex and dynamic, and involves both a large variety of structural conformations and a dynamic nature of these structures as key element in their function. Protein conformations of crystallized samples are known, but conformations in liquid still remain elusive. This issue can only be partly answered by cryo-EM techniques using rapid freezing of different dynamics states,73 while LP-EM could provide unique nanoscale of structures in liquid state in future.
LP-EM has enabled remarkable discoveries at the macromolecular level in a liquid environment, but its use for the study of complex protein–protein interaction processes has not yet been explored. Although imaging live cells with electrons is highly challenging,52 many relevant biochemical processes take place in the extracellular space. Typically, proteins do not act in isolation when performing their functions in vivo, but rather assemble in macromolecular complexes that interact driven by physicochemical parameters. LP-EM coupled with in-liquid immunolabeling techniques has the potential to become a powerful tool for the study of protein interactions at the extracellular level, with a significant impact on biomedical research. However, several key innovations are needed to achieve near-native molecular dynamics visualization under physiologically relevant conditions.
Crucial effort should be dedicated to the avoidance of radiation damage,5 with different approaches already having promising potential. For instance, the dose tolerance can be increased by using graphene encapsulation, due to the ability of graphene to quench an excited state and conduct charge away from the sample.21,22 It is thus of key importance to implement reliable and stable methods for the creation of graphene liquid cells (GLCs).74 In addition, the electron flux needs to be minimized so as to allow reversible processes to equilibrate, for example, after the creation of charge pairs.22,32 Additionally, the thickness of the sample must be minimized to limit the amount of inelastically scattered electrons in the sample, as those are the main cause of damage. A balance must be found between knock-on damage and the creation of radicals by optimizing the beam energy, combined with penetration depth of the beam in the sample. Radical scavengers, such as vitamin C and many others, can be added to increase the dose tolerance.
On the other hand, the detection needs to be optimized as much as possible, preferably by several orders of magnitude. To achieve this improvement, advanced LP-EM technology for ultralow-dose detection using direct electron detectors can be used. The phase contrast signal can be increased in the nanometer resolution range using phase plate technology.75 Further exploration is required for a new option involving differential phase contrast STEM.76 To improve the resolution-electron dose balance even further, intelligent acquisition schemes need to be applied, instead of scanning every pixel in an image or image sequence. One approach is to apply a higher dose to edges of structures containing features, rather than the surrounding area without features, using so-called adaptive scanning.77 A second strategy is randomly scanning sparsely distributed pixels and reconstructing an image via various methods, including in-painting techniques, for example.78,79
By combining state-of-the-art technologies already existing in the electron microscopy community, LP-EM can be expected to safely study physiological processes occurring in proteins, DNA, bacterial cells, mammalian cells, tissue, bone, and more in the near future. A unique nanoscale view on the dynamic biological world will help scientists better understand protein function, virus–cell interactions, bone mineralization, and find clues toward developing better medication. LP-EM adds a unique view compared with other techniques, such as super-resolution microscopy and cryo-TEM, which is needed to study the highly complex molecular world of biology from different perspectives.
References
N. de Jonge, F.M. Ross, Nat. Nanotechnol. 6, 695 (2011).
N. de Jonge, D.B. Peckys, G.J. Kremers, D.W. Piston, Proc. Natl. Acad. Sci. U.S.A. 106, 2159 (2009).
F.M. Ross, Liquid Cell Electron Microscopy (Cambridge University Press, Cambridge, UK, 2017).
N. de Jonge, L. Houben, R.E. Dunin-Borkowski, F.M. Ross, Nat. Rev. Mater. 4, 61 (2019).
H. Wu, H. Friedrich, J.P. Patterson, N. Sommerdijk, N. de Jonge, Adv. Mater. 32, 2001582 (2020).
K. He, T. Shokuhfar, R. Shahbazian-Yassar, J. Phys. Condens. Matter 31, 103001 (2019).
D.F. Parsons, V.R. Matricardi, R.C. Moretz, J.N. Turner, Adv. Biol. Med. Phys. 15, 161 (1974).
H. Sugi, T. Akimoto, K. Sutoh, S. Chaen, N. Oishi, S. Suzuki, Proc. Natl. Acad. Sci. U.S.A. 94, 4378 (1997).
G.D. Danilatos, in Advances in Electronics and Electron Physics (Academic Press, 1988), vol. 71, p. 109.
D.L. Stokes, Principles and Practice of Variable Pressure/Environmental Scanning Electron Microscopy (VP-ESEM) (Wiley, Chichester, UK, 2008).
A. Bogner-Van De Moortele, G. Thollet, D. Basset, P.-H. Jouneau, C. Gauthier, Ultramicroscopy 104, 290 (2005).
D.B. Peckys, J.P. Baudoin, M. Eder, U. Werner, N. de Jonge, Sci. Rep. 3, 2626 (2013).
S. Thiberge, A. Nechushtan, D. Sprinzak, O. Gileadi, V. Behar, O. Zik, Y. Chowers, S. Michaeli, J. Schlessinger, E. Moses, Proc. Natl. Acad. Sci. U.S.A. 101, 3346 (2004).
N. de Jonge, D.B. Peckys, G.M. Veith, S. Mick, S.J. Pennycook, C.S. Joy, Microsc. Microanal. 13 (Suppl. 2), 242 (2007).
M.J. Williamson, R.M. Tromp, P.M. Vereecken, R. Hull, F.M. Ross, Nat. Mater. 2, 532 (2003).
D.B. Peckys, G.M. Veith, D.C. Joy, N. de Jonge, PLoS One 4, e8214 (2009).
H. Nishiyama, M. Suga, T. Ogura, Y. Maruyama, M. Koizumi, K. Mio, S. Kitamura, C. Sato, J. Struct. Biol. 169, 438 (2010).
J. Park, H. Park, P. Ercius, A.F. Pegoraro, C. Xu, J.W. Kim, S.H. Han, D.A. Weitz, Nano Lett. 15, 4737 (2015).
M. Textor, N. de Jonge, Nano Lett. 18, 3313 (2018).
S.M. Ghodsi, C.M. Megaridis, R. Shahbazian-Yassar, T. Shokuhfar, Small Methods 3, 1900026 (2019).
H. Cho, M.R. Jones, S.C. Nguyen, M.R. Hauwiller, A. Zettl, A.P. Alivisatos, Nano Lett. 17, 414 (2017).
S. Keskin, N. de Jonge, Nano Lett. 18, 7435 (2018).
W.J. Dearnaley, B. Schleupner, A.C. Varano, N.A. Alden, F. Gonzalez, M.A. Casasanta, B.E. Scharf, M.J. Dukes, D.F. Kelly, Nano Lett. 19, 6734 (2019).
G. Marchello, C. De Pace, N. Wilkinson, L. Ruiz-Perez, G. Battaglia, preprint, arXiv:1907.03348 (2019).
K. Masenelli-Varlot, A. Malchere, J. Ferreira, H. Heidari Mezerji, S. Bals, C. Messaoudi, S. Marco Garrido, Microsc. Microanal. 20, 366 (2014).
N. de Jonge, L. Houben, R.E. Dunin-Borkowski, F.M. Ross, Nat. Rev. Mater. 4, 61 (2019).
T.J. Woehl, P. Abellan, J. Microsc. 265, 135 (2017).
A. Hoenger, C. Bouchet-Marquis, Adv. Protein Chem. Struct. Biol. 82, 67 (2011).
J. Hermannsdörfer, V. Tinnemann, D.B. Peckys, N. de Jonge, Microsc. Microanal. 20, 656 (2016).
V.R. Matricardi, R.C. Moretz, D.F. Parsons, Science 177, 268 (1972).
U.M. Mirsaidov, H. Zheng, Y. Casana, P. Matsudaira, Biophys. J. 102, L15 (2012).
S. Keskin, S. Besztejan, G. Kassier, S. Manz, R. Bucker, S. Riekeberg, H.K. Trieu, A. Rentmeister, R.J. Miller, J. Phys. Chem. Lett. 6, 4487 (2015).
M. Wang, C. Park, T. Woehl, Microsc. Microanal. 25, 23 (2019).
N. de Jonge, Ultramicroscopy 187, 113 (2018).
T.H. Moser, T. Shokuhfar, J.E. Evans, Micron 117, 8 (2019).
J.P. Baudoin, J.R. Jinschek, C.B. Boothroyd, R.E. Dunin-Borkowski, N. de Jonge, Microsc. Microanal. 19, 814 (2013).
M.F. Hohmann-Marriott, A.A. Sousa, A.A. Azari, S. Glushakova, G. Zhang, J. Zimmerberg, R.D. Leapman, Nat. Methods 6, 729 (2009).
M. Elbaum, S.G. Wolf, L. Houben, MRS Bull. 41, 542 (2016).
D.B. Peckys, P. Mazur, K.L. Gould, N. de Jonge, Biophys. J. 100, 2522 (2011).
I.N. Dahmke, A. Verch, J. Hermannsdörfer, D.B. Peckys, R.S. Weatherup, S. Hofmann, N. de Jonge, ACS Nano 11, 11108 (2017).
J.E. Evans, K.L. Jungjohann, P.C.K. Wong, P.L. Chiu, G.H. Dutrow, I. Arslan, N.D. Browning, Micron 43, 1085 (2012).
C. Wang, Q. Qiao, T. Shokuhfar, R.F. Klie, Adv. Mater. 26, 3410 (2014).
M.J. Dukes, R. Thomas, J. Damiano, K.L. Klein, S. Balasubramaniam, S. Kayandan, J.S. Riffle, R.M. Davis, S.M. McDonald, D.F. Kelly, Microsc. Microanal. 20, 338 (2014).
C. Wadell, S. Inagaki, T. Nakamura, J. Shi, Y. Nakamura, T. Sannomiya, ACS Nano 11, 1264 (2017).
T. Yamazaki, Y. Kimura, P.G. Vekilov, E. Furukawa, M. Shirai, H. Matsumoto, A.E.S. Van Driessche, K. Tsukamoto, Proc. Natl. Acad. Sci. U.S.A. 114, 2154 (2017).
A.C. Varano, A. Rahimi, M.J. Dukes, S. Poelzing, S.M. McDonald, D.F. Kelly, Chem. Commun. 51, 16176 (2015).
H. Wang, B. Li, Y.J. Kim, O.H. Kwon, S. Granick, Proc. Natl. Acad. Sci. U.S.A. 117, 1283 (2020).
K.L. Liu, C.C. Wu, Y.J. Huang, H.L. Peng, H.Y. Chang, P. Chang, L. Hsu, T.R. Yew, Lab Chip 8, 1915 (2008).
N. Mohanty, M. Fahrenholtz, A. Nagaraja, D. Boyle, V. Berry, Nano Lett. 11, 1270 (2011).
T. Prozorov, T.P. Almeida, A. Kovacs, R.E. Dunin-Borkowski, J. R. Soc. Interface 14, 20170464 (2017).
K. Koo, K.S. Dae, Y.K. Hahn, J.M. Yuk, Nano Lett. 20, 4708 (2020).
N. de Jonge, D.B. Peckys, ACS Nano 10, 9061 (2016).
L. Reimer, H. Kohl, Transmission Electron Microscopy: Physics of Image Formation (Springer, New York, 2008).
B.N. Giepmans, T.J. Deerinck, B.L. Smarr, Y.Z. Jones, M.H. Ellisman, Nat. Methods 2, 743 (2005).
T. Ando, S.P. Bhamidimarri, N. Brending, H. Colin-York, L. Collinson, N. de Jonge, P.J. de Pablo, E. Debroye, C. Eggeling, C. Franck, M. Fritzsche, H. Gerritsen, B.N.G. Giepmans, K. Grunewald, J. Hofkens, J.P. Hoogenboom, K.P.F. Janssen, R. Kaufman, J. Klumpermann, N. Kurniawan, J. Kusch, N. Liv, V. Parekh, D.B. Peckys, F. Rehfeldt, D.C. Reutens, M.B.J. Roeffaers, T. Salditt, I.A.T. Schaap, U.S. Schwarz, P. Verkade, M.W. Vogel, R. Wagner, M. Winterhalter, H. Yuan, G. Zifarelli, J. Phys. D Appl. Phys. 51, 443001 (2018).
Y. Maruyama, T. Ebihara, H. Nishiyama, M. Suga, C. Sato, J. Struct. Biol. 180, 259 (2012).
D.B. Peckys, N. de Jonge, Microsc. Microanal. 20, 189 (2014).
D.B. Peckys, D. Hirsch, T. Gaiser, N. de Jonge, Mol. Med. 25, 42 (2019).
D.B. Peckys, C. Stoerger, L. Latta, U. Wissenbach, V. Flockerzi, N. de Jonge, J. Struct. Biol. 199, 102 (2017).
D. Alansary, D.B. Peckys, B.A. Niemeyer, N. de Jonge, J. Cell Sci. 133, 240358 (2020).
D.B. Peckys, U. Korf, S. Wiemann, N. de Jonge, Mol. Biol. Cell 28, 3193 (2017).
T. Yamazawa, N. Nakamura, M. Sato, C. Sato, Microsc. Res. Tech. 79, 1179 (2016).
N. Memtily, T. Okada, T. Ebihara, M. Sato, A. Kurabayashi, M. Furihata, M. Suga, H. Nishiyama, K. Mio, C. Sato, Int. J. Oncol. 46, 1872 (2015).
M.H. Nielsen, S. Aloni, J.J. De Yoreo, Science 345, 1158 (2014).
P.J. Smeets, K.R. Cho, R.G. Kempen, N.A. Sommerdijk, J.J. De Yoreo, Nat. Mater. 14, 394 (2015).
Z. Liu, Z. Zhang, Z. Wang, B. Jin, D. Li, J. Tao, R. Tang, J.J. De Yoreo, Proc. Natl. Acad. Sci. 117, 3397 (2020).
A. Dey, P.H. Bomans, F.A. Muller, J. Will, P.M. Frederik, G. de With, N.A. Sommerdijk, Nat. Mater. 9, 1010 (2010).
X.Y. Wang, J. Yang, C.M. Andrei, L. Soleymani, K. Grandfield, Commun. Chem. 1, 80 (2018).
C. Sato, D. Yamazaki, M. Sato, H. Takeshima, N. Memtily, Y. Hatano, T. Tsukuba, E. Sakai, Sci. Rep. 9, 7352 (2019).
F. Balzarotti, Y. Eilers, K.C. Gwosch, A.H. Gynna, V. Westphal, F.D. Stefani, J. Elf, S.W. Hell, Science 355, 606 (2017).
L.F. Kourkoutis, J.M. Plitzko, W. Baumeister, Annu. Rev. Mater. Res. 42, 33 (2012).
J. Lotze, U. Reinhardt, O. Seitz, A.G. Beck-Sickinger, Mol. Biosyst. 12, 1731 (2016).
K. Murata, M. Wolf, Biochim. Biophys. Acta 1862, 324 (2018).
P.M.G. van Deursen, R.I. Koning, V. Tudor, M.-A. Moradi, J.P. Patterson, A. Kros, N.A. Sommerdijk, A.J. Koster, G.F. Schneider, Adv. Funct. Mater. 30, 1904468 (2020).
R. Danev, R.M. Glaeser, K. Nagayama, Ultramicroscopy 109, 312 (2009).
I. Lazic, E.G.T. Bosch, S. Lazar, Ultramicroscopy 160, 265 (2016).
T. Dahmen, M. Engstler, C. Pauly, P. Trampert, N. de Jonge, F. Mucklich, P. Slusallek, Sci. Rep. 6, 25350 (2016).
A. Stevens, H. Yang, L. Carin, I. Arslan, N.D. Browning, Microscopy 63, 41 (2013).
A. Beche, B. Goris, B. Freitag, J. Verbeeck, Appl. Phys. Lett. 108, 93103 (2016).
Acknowledgments
D.B.P. was supported by the DFG SFB1027 (Project No. C7). N.d.J. acknowledges E. Arzt for his support through INM.
Appendix
Appendix

 Diana B. Peckys has been a research scientist in the Department of Biophysics at Saarland University, Germany, since 2016. She received her master's degree in 1994, and her PhD degree in 1998 in human biology from the University of Marburg, Germany. She worked at Philips Medical Systems, The Netherlands, from 2000 to 2005, Oak Ridge National Laboratory and the University of Tennessee from 2005 to 2010, and as a research instructor at Vanderbilt University School of Medicine from 2007 to 2011. She was a research scientist at the INM–Leibniz Institute for New Materials, Germany, from 2012 to 2015. Her research includes co-pioneering liquid phase electron microscopy for biological samples, and focuses on cell biology and cancer. Peckys can be reached by email at diana.peckys@uks.eu.
Diana B. Peckys has been a research scientist in the Department of Biophysics at Saarland University, Germany, since 2016. She received her master's degree in 1994, and her PhD degree in 1998 in human biology from the University of Marburg, Germany. She worked at Philips Medical Systems, The Netherlands, from 2000 to 2005, Oak Ridge National Laboratory and the University of Tennessee from 2005 to 2010, and as a research instructor at Vanderbilt University School of Medicine from 2007 to 2011. She was a research scientist at the INM–Leibniz Institute for New Materials, Germany, from 2012 to 2015. Her research includes co-pioneering liquid phase electron microscopy for biological samples, and focuses on cell biology and cancer. Peckys can be reached by email at diana.peckys@uks.eu.
 Elena Macías-Sánchez is an assistant professor at the Radboud University Medical Center, The Netherlands. She completed postdoctoral research in the Department of Biomaterials at the Max Planck Institute of Colloids and Interfaces, Germany, from 2017 to 2020. She received her master's degree in genetics and evolution, and her PhD degree in biomineralization from the University of Granada, Spain, in 2017. Her research includes the study of the molecular mechanisms of bone mineralization, both in health and disease. Her current research focuses on a correlative approach combining liquid phase electron microscopy for the study of the mineralization processes in situ. Macías-Sánchez can be reached by email at elena.macias@radboudumc.nl.
Elena Macías-Sánchez is an assistant professor at the Radboud University Medical Center, The Netherlands. She completed postdoctoral research in the Department of Biomaterials at the Max Planck Institute of Colloids and Interfaces, Germany, from 2017 to 2020. She received her master's degree in genetics and evolution, and her PhD degree in biomineralization from the University of Granada, Spain, in 2017. Her research includes the study of the molecular mechanisms of bone mineralization, both in health and disease. Her current research focuses on a correlative approach combining liquid phase electron microscopy for the study of the mineralization processes in situ. Macías-Sánchez can be reached by email at elena.macias@radboudumc.nl.
 Niels de Jonge has been a senior group leader at the INM–Leibniz Institute for New Materials, Germany, since 2012, and an honorary professor of physics at Saarland University, Germany. He received his master's degree in physics from the University of Amsterdam, The Netherlands, in 1994, and his PhD degree in biophysics from the University of Freiburg, Germany, in 1999. He subsequently worked as a senior scientist at Philips Research, The Netherlands, from 2000 to 2005. He studied liquid phase electron microscopy at Oak Ridge National Laboratory from 2005 to 2010, while also serving as an assistant professor at Vanderbilt University School of Medicine from 2007 to 2011. De Jonge can be reached by email at Niels.deJonge@leibniz-inm.de.
Niels de Jonge has been a senior group leader at the INM–Leibniz Institute for New Materials, Germany, since 2012, and an honorary professor of physics at Saarland University, Germany. He received his master's degree in physics from the University of Amsterdam, The Netherlands, in 1994, and his PhD degree in biophysics from the University of Freiburg, Germany, in 1999. He subsequently worked as a senior scientist at Philips Research, The Netherlands, from 2000 to 2005. He studied liquid phase electron microscopy at Oak Ridge National Laboratory from 2005 to 2010, while also serving as an assistant professor at Vanderbilt University School of Medicine from 2007 to 2011. De Jonge can be reached by email at Niels.deJonge@leibniz-inm.de.
Rights and permissions
About this article

Cite this article
Peckys, D.B., Macías-Sánchez, E. & de Jonge, N. Liquid phase electron microscopy of biological specimens. MRS Bulletin 45, 754–760 (2020). https://doi.org/10.1557/mrs.2020.225
Published:
Issue Date:
DOI: https://doi.org/10.1557/mrs.2020.225