
Abstract
Carbon-coated silicon nanowires (C-Si NWs) were prepared as anodes for lithium-ion batteries (LIBs). The C-Si NWs were synthesized using a simple and effective fabrication strategy via magnesiothermic reduction. The synthesis sequence of carbon coating before the chemical etching of the reduced Si NWs/MgO composite was found to be critical for improved battery performance. In addition, carbon coating was found to help to stabilize the solid electrolyte interphase layer during battery cycling, which is important to realize the benefits of Si-based LIBs. This synthesis method provides an efficient route to synthesizing high-performance Si electrodes via magnesiothermic reduction.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lithium-ion batteries (LIBs) have received considerable attention as the most advanced secondary batteries, because of their high-energy densities per volume and per weight. Over the past two decades, there has been significant research on LIBs to improve the energy density, cycle performance and rate capability for high-power and high-energy applications such as power tools, mobile electronics, and electric vehicles.[1] In addition, LIBs are favorable candidates for various stationary power storage applications such as renewable energy.[2] The performance of LIBs is mostly dependent on the characteristics of the electrode materials. Silicon (theoretical capacity is 4200 mAh/g based on Li4.4Si compared with 372 mAh/g for graphite[3]) is the most promising anode materials that could replace the current graphite electrodes. However, the increased Li richness in Si anodes during cycling processes results in breaking the Si-Si bonds due to large volume changes of Si up to 400%.[4,5] This repeated volume changes in the process of lithiation/delithiation causes pulverization and short battery life. For the same reason, as the cycle progresses, the solid electrolyte interphase (SEI) films on the surface of the electrodes become unstable and increase in thickness. The thick SEI films increase the Li diffusion resistance, and consumption of a large amount of cathode’s Li-ions.[6] Unless these technical issues are overcome, the realization of the benefits of Si-based LIBs will continue to be elusive.
Progress in nanofabrication and nanomaterials have made significant breakthroughs to tackle the challenges of Si-based LIBs. Recent experimental and theoretical studies suggest several advantages of nanostructured electrode materials. These include: (1) increased surface area, resulting in higher lithium-ion flux across the electrode/electrolyte interface, (2) better resilience to mechanical failure, and (3) higher lithium diffusion and electron transfer rates.[7–10] Studies have proposed Si nanostructures as anodes in LIBs with different morphologies such as nanowires,[6] hollow structures,[11] porous structures,[12] and composites.[13] Silicon nanowires are particularly promising due to the short Li diffusion distance and direct one-dimensional (1D) electron transport path.[14,15]
In addition, thin carbon films have been applied to active materials as a protective layer to improve the mechanical and electrochemical characteristics of Si anodes. For example, Yoshio et al. showed that carbon films on Si powders suppress further decomposition of the electrolyte, resulting in improved electrochemical performance.[16] Also, carbon films on Si NWs improved the electrical conductivity and accommodated the volume change of the Si NWs during cycling processes.[17]
Nanostructured Si has been synthesized using various techniques including chemical etching of a silicon wafer,[18] low-pressure chemical vapor deposition (LP-CVD) using silane (SiH4) as a precursor,[19] thermal vapor deposition,[20] laser ablation,[21] and magnesiothermic reduction of silica.[22] Unlike the techniques which involve complicated processes and the use of toxic precursors such as SiH4, magnesiothermic reduction has a great potential as an economical and environmentally friendly route for large-scale production of nanostructured Si. Silica (SiO2) can be reduced to crystalline Si through a chemical route in which magnesium is typically used as the reducing agent. The main advantage of the magnesiothermic reduction technique is to retain the morphology of silica reactants in the final Si product with the same geometric features. SiO2 is abundant; replicable silica-forming micro-organisms can be easily acquired in nature such as diatoms and rice husk ash for biologic silica templates.[22,23] In addition, the fabrication methods for synthetic SiO2 are well established. One drawback is the incomplete magnesiothermic reduction of SiO2 into Si due to the formation of unavoidable silicon-bearing gas species byproducts such as magnesium silicide.[24] In addition, in case of carbon coating, typically, it is applied to Si materials after etching the byproducts in the composite when it comes to magnesiothermic reduction.[25]
In this work, we present the synthesis method and electrochemical performance of carbon-coated silicon nanowires (C-Si NWs) as anodes for LIBs. We demonstrate that the synthesis sequence influences not only the structure of the final product but also the performance of the LIBs. Si NWs were prepared by magnesiothermic reduction of SiO2 NWs and electrically interconnected through carbon films with void spaces. To ensure porous silicon NWs with controlled morphology, the etching of the byproduct in the composite was conducted after the carbon coating. Carbon coating also helps to stabilize the SEI layer during battery cycling and improve the electrical conductivity, and thus leading to a fast and efficient electron transfer pathway. The produced C-Si NWs exhibited improved electrochemical performance.
Experimental
Synthesis of C-Si NWs
A magnesiothermic reduction of silica was conducted to produce silicon nanostructures, which can be described by the following chemical reaction:
The silica precursor including nanoparticles (NPs; used for comparison, purchased from US Research Nanomaterials, Inc., USA) and nanowires (supplied by DaeBaek, LLC., USA) were mixed with magnesium powder in a mass ratio of 1:1 for chemical reduction. The mixed powders were sealed in a stainless-steel container under high-purity argon (>99.995%) and placed into a tube furnace. The tube furnace was heated to 675 °C and held constant for 2 h under argon. The evaporated magnesium reacts with silica and forms Si/ MgO composites. Then, the chamber was left to cool down to room temperature. As a result, the yellow-brown powder Si/MgO composite was collected, and the sample was placed in an LP-CVD reactor under acetylene (C2H2) gas diluted by argon for the carbon coating. The gas flow ratio of C2H2 and argon was fixed at 1:10 for 20 min at a temperature of 620 °C. Subsequently, the resulting powder was stirred in 1 M HCl solution for 2 h to remove the byproducts including MgO and Mg2Si. After washing with distilled water and dried in a vacuum oven overnight at 60 °C, the final product was Si NWs coated with carbon film.
Assembly and characterizations of LIBs
The weight of the active materials was obtained by measuring the weight difference after the carbon deposition with a Sartorius CPA225D micro-balance (0.01 mg resolution). The typical mass loading of Si NWs anode was 1.5 mg/cm. The prepared C-Si NWs materials were used for anodes without further treatment. The mixture of hybrid materials, carbon black, and poly(acrylic acid) (PAA) binder (60:20:20 by weight) in N-methyl-2-pyrrolidinone was prepared to form a slurry. The slurry was coated onto a copper foil using a film applicator and dried overnight in a vacuum oven at 100 °C. The coin-cell (CR2032) with the lithium metal foil and the reference electrode was assembled in a glove box under an argon atmosphere with oxygen and moisture levels below 1 ppm. The electrolyte consists of 1 M Lithium hexafluorophosphate (LiPF6) solution in a carbonate mixture (ethylene carbonate/diethyl carbonate = 50:50 by volume). The coin cells were cycled using a Land Battery Test System in a galvanostatic mode, for their electrochemical performance at room temperature.
Results and discussion
In this study, the charge and discharge processes are considered as lithiation and delithiation of Si, respectively. The specific energy density was based on the weight of Si and C materials in the hybrid. For comparison, commercial SiO2 NPs were also used to prepare the C-Si NPs anodes. All electrodes were initially run at a current rate of 0.05 C (40 mA/g) with a voltage ranging from 0.01 to 2.0 V versus Li+/Li for the first 2 cycles. Then, the galvanostatic cycling was conducted at a current rate of 0.1 C (200 mA/g). The battery performance was explored with Si products reduced from silica materials. The magnesiothermic reduction of silica using magnesium powder as a reducing agent produces a MgO/Si composite: 34.9% of Si and 64.1% of MgO by volume.
The composite contained high volume fraction of MgO. Chemical etching after the magnesiothermic reduction was required to remove the byproducts such as MgO and Mg2Si. The resultant Si became highly porous due to the dissolved MgO in the composite. For utilizing the porous nature of the reduced Si for efficient volume accommodation, thin carbon films were deposited on the resultant Si (after chemical etching) and MgO/Si composite. For a C-Si NPs-a, carbon coating was conducted before the chemical etching. As a control, another sample, C-Si NPs-b was made with carbon coating after the chemical etching. The electrochemical performance of both C-Si NPs was compared. As shown in Fig. 1, C-Si NPs-a exhibited improved cycle performance, retaining 71% of its initial capacity after 30 cycles, while the control sample, C-Si NPs-b showed 18%. The carbon coating before the chemical etching was found to be a crucial factor to improve battery perforce. The pore may have remained in the C-Si NPs-a after chemical etching of MgO in the composite, which accommodated the volume changes of Si during battery cycling.

The reversible delithiation capacity of C-Si NPs-a (chemically etched after carbon coating) and C-Si NPs-b (chemically etched before carbon coating) between 0.01 and 2.0 V at a current rate of 0.1 C (or 200 mA/g).
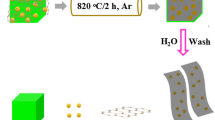
To further improve the electrochemical performance of Si-based electrodes with the magnesiothermic reduction, we used SiO2 NWs and prepared C-Si NWs with the aforementioned recipe. Figure 2 illustrates the procedure for fabricating C-Si NWs. As received SiO2 NWs were reduced to Si NWs via magnesiothermic reduction, and the resultant powder was coated with thin carbon films using LP-CVD. Subsequently, the remaining SiO2 and by-products including MgO and Mg2Si have etched away with HCl. The final product was carbon coated Si NWs (C-Si NWs).

Schematic illustration of the synthesis of Si NWs with carbon coating. The fabrication includes magnesiothermic reduction of SiO2 and carbon coating through chemical vapor deposition, followed by the chemical etching of the by-products.
To examine the structure, morphology and composition of C-Si NWs, scanning electron microscopy (SEM) analysis was performed using a JEOL JSM-7500F SEM. Figure 3(a) shows the reduced Si nanowires with carbon coating and a higher magnification scan is obtained for a single C-Si NW. The images revealed that Si NWs maintained the original structure, which is consistent with previous research on magnesiothermic reduction of silica. The carbon coating, which covered the surface of the Si NWs, formed continuous films to enhance electrical pathways.

(a) SEM image of a C-Si NW and inset showing low magnification SEM image for C-Si NWs, (b) elemental peaks from the line scan across the single C-Si NW, (c) and (d) elemental maps of C and Si, respectively.
Figure 3(b-d) reveals the energy dispersive spectroscopic (EDS) analysis performed on a single C-Si NW, showing strong Si and C signals. The EDS analysis was performed using both line scan and 2D mapping. The line scan showed an increase of Si signal towards the center of the nanowire, while it showed a slightly decreasing signal for C. Moreover, the line scan provides an estimate of the thickness of the carbon coating to be about 50 nm. The elemental maps of silicon and carbon on the hybrid material indicated that Si NW was completely covered with the carbon films.
The crystallographic structure of silica and Si NWs were investigated through x-ray diffraction method (XRD, Bruker-AXS D8 VARIO with Cu Kα x-ray). The silica nanowires exhibited a broad peak that resulted from their amorphous structure, whereas three sharp peaks appeared in the XRD pattern after the magnesiothermic reduction, as shown in Fig. S1. The strong peaks indicated the crystalline structure of the reduced Si NWs. The broad peak in the Si NWs samples indicates that amorphous silica still exists after the reduction treatment. However, by-products such as MgO and Mg2Si were completely removed after the reduction. Figure S2 shows the thermogravimetric (TGA) analysis, which was conducted to determine the content of carbon in the hybrid structure. The C-Si NWs were heated in alumina pan at a heating rate of 10°C/min under air for a maximum temperature of 700 °C. Since Si oxidation is expected to happen rapidly above 750 °C,[26] the weight loss of the sample is mainly attributed to the combustion of carbon below this temperature. The TGA revealed that the content of carbon in the hybrid was about 3wt %.
Figure 4(a) shows the cycling performance of C-Si NPs and C-Si NWs electrodes during prolonged cycling. The C-Si NWs exhibited improved electrochemical performance including their specific capacity and stability, compared with C-Si NPs. The specific capacity in the first cycle of C-Si NPs and C-Si NWs were 1342 and 1257 mAh/g, respectively. After 40 cycles, the capacity of each sample remained at 733 and 940 mAh/g, respectively. The reversible capacities of both samples remained higher than that of graphite. C-Si NPs showed distinct fading of charge/discharge capacity in the first few cycles, while the C-Si NWs samples exhibited a much slower decay of capacity with cycling. The capacity loss may be ascribed to the unstable SEI layer formation, which increased the diffusion resistance and consumption of large amounts of Li-ions during cycling.

Electrochemical measurements of C-Si NWs anodes. All the specific capacities of the anodes are based on the total mass of the active materials (Si and C in the composite materials). (a) Reversible delithiation capacity for the first 40 galvanostatic cycles of carbon coated Si NWs and NPs at a current rate of 0.1 C, (b) Reversible delithiation capacity of C-Si NWs at different current rates, (c) Charge-discharge curves of C-Si NPs and C-Si NWs between 0.0 and 2.0 V at a current rate of 0.1 C and (d) Differential capacity (dQ/dV) curves of C-Si NWs between 0.0 and 2.0 V at a current rate of 0.1 C.
The rate performance of C-Si NWs at various current rates was tested every 10 cycles, as shown in Fig. 4(b). The cells were cycled with a low current rate of 0.1 C in the first 10 cycles, and the current rates were increased gradually. The reversible capacities were measured as 1334 mAh/g at 0.1 C, 1102 mAh/g at 0.5 C, 755 mAh/g at 1 C, and 435 mAh/g at 2 C, which is due to increased resistance at the interface between the electrolyte and the active materials at a higher current rate. The current rate was lowered back to 0.5 C after 40 cycles and the reversible capacity was 989 mAh/g. The improved electrochemical performance of C-Si NWs may be attributed to the void space produced during the chemical etching process. The etching operation after the carbon deposition effectively produced void space for Si to expand freely without confinement.
Figure 4(c) shows the initial charge/discharge voltage profiles of Si NWs and Si NPs electrodes at 0.1 C. The Coulombic efficiencies in the first cycle for Si NWs and NPs were 77% and 90%, respectively. The irreversible capacity loss in the first cycle was mainly attributed to the SEI formation and irreversible reaction with the remaining oxygen-containing functional group. There was a sharp drop in the voltage to about 0.1 V and a long plateau at 0.1–0.01 V forming an amorphous LixSi phase. No other plateau that corresponded to the Li intercalation into the carbon was observed. A broad plateau at around 0.5 V can be observed in the discharging process for electrodes, corresponding to the dealloying reaction of the Si host with Li. Fig. 4(d) illustrates the corresponding differential profiles of C-Si NWs. The differential capacity (dQ/dV) was acquired by numerical differentiation of the galvanostatic cycling data. It shows a sharp anodic peak at 0.04 V in the first charge curve, which was attributed to the coexistence of two phase regions of amorphous LixSi and crystalline Si. In the discharging curve, a broad and a sharp peak at around 0.3 and 0.45 V were related to the Li de-alloying reaction forming amorphous Si.
In summary, the C-Si NWs composite using both magnesio-thermic reduction and LP-CVD techniques delivered promising electrochemical performance due to their unique 1D porous structure. A specific capacity of 1000 mAh/g at a current rate of 0.1 C was obtained with 25% irreversible capacity loss after 40 cycles. The synthetic procedure of the C-Si NWs is fairly simple, and scalable. Due to the unique morphology and structure of Si NWs, they have a great potential as anodes for LIBs. The careful control of synthetic procedure of carbon coating and chemical etching ofthe Si/C composite contributed to the higher electrochemical performance. Carbon coating before the chemical etching results in porous structure and the void space allowed the silicon expansion appropriately during charging/discharging. Therefore, the carbon film played a vital role in both as a protective film to suppress the SEI formation on the surface of Si and a clamping layer in order to accommodate the significant volumetric variation while using the LIB. In addition, the whole surface of the Si NWs was covered with carbon films, which protected the Si NWs from direct contact with the electrolyte, thus stabilize the growth of SEI.
Conclusions
The electrochemical performance of a C-Si NWs was investigated for anodes in LIBs. SiO2 was successfully reduced into Si using magnesiothermic reduction, and Si NWs were coated with carbon films using the LP-CVD method. The electrochemical tests showed that C-Si NWs anodes could be cycled up to 40 cycles with a reversible specific capacity around 1000 mAh/g, which was higher than that of C-Si NPs anodes. We demonstrated that the void space in C-Si NWs effectively accommodates strain of the Si NWs during cycling. Further studies would accomplish improved cycle performance of C-Si NWs by controlling the SEI formation with structural modification of the Si. The material synthesis and electrode fabrication processes are relatively simple, scalable, highly reproducible, and compatible with slurry coating manufacturing technology. The proposed fabrication approach gave an insight on how to construct SiO2-based electrodes through a chemical reduction as anode for enhanced Li-ion battery performance.
References
J.B. Goodenough and A. Manthiram: A perspective on electrical energy storage. MRS Commun. 4, 135 (2014).
B. Diouf and R. Pode: Potential of lithium-ion batteries in renewable energy. Renew. Energy 76, 375 (2015).
T.S.D. Kumari, R. Surya, A.M. Stephan, D. Jeyakumar, and T. Prem Kumar: High-capacity potato peel-shaped graphite for lithium-ion batteries. MRS Commun. 1, 41 (2011).
U. Kasavajjula, C. Wang, and A.J. Appleby: Nano- and bulk-silicon-based insertion anodes for lithium-ion secondary cells. J. Power Sources 163, 1003 (2007).
S. Kim and H.-Y.S. Huang: Mechanical stresses at the cathode-electrolyte interface in lithium-ion batteries. J. Mater. Res. 31, 3506 (2016).
C.K. Chan, H. Peng, G. Liu, K. McIlwrath, X.F. Zhang, R.A. Huggins, and Y. Cui: High-performance lithium battery anodes using silicon nanowires. Nat Nano 3, 31 (2008).
X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B.W. Sheldon, and J. Wu: Silicon-based nanomaterials for lithium-ion batteries: a review. Adv. Energy Mater. 4, 1300882 (2014).
Y. Wang and G. Cao: Developments in nanostructured cathode materials for high-performance lithium-ion batteries. Adv. Mater. 20, 202251 (2008).
L. Mai, X. Xu, L. Xu, C. Han, and Y. Luo: Vanadium oxide nanowires for Li-ion batteries. J. Mater. Res. 26, 2175 (2011).
B. Liu, D. Deng, J.Y. Lee, and E.S. Aydil: Oriented single-crystalline TiO2 nanowires on titanium foil for lithium ion batteries. J. Mater. Res. 25, 1588 (2010).
X. Huang, J. Yang, S. Mao, J. Chang, P.B. Hallac, C.R. Fell, B. Metz, J. Jiang, P.T. Hurley, and J. Chen: Controllable synthesis of hollow Si anode for long-cycle-life lithium-ion batteries. Adv. Mater. 26, 4326 (2014).
M. Ge, J. Rong, X. Fang, A. Zhang, Y. Lu, and C. Zhou: Scalable preparation of porous silicon nanoparticles and their application for lithium-ion battery anodes. Nano Res. 6, 174 (2013).
B. Wang, X. Li, X. Zhang, B. Luo, M. Jin, M. Liang, S.A. Dayeh, S. T. Picraux, and L. Zhi: Adaptable silicon-carbon nanocables sandwiched between reduced graphene oxide sheets as lithium ion battery anodes. ACS Nano 7, 1437 (2013).
K. Peng, J. Jie, W. Zhang, and S.-T. Lee: Silicon nanowires for rechargeable lithium-ion battery anodes. Appl. Phys. Lett. 93, 33105 (2008).
C.K. Chan, R.N. Patel, M.J. O'Connell, B.A. Korgel, and Y. Cui: Solution-grown silicon nanowires for lithium-ion battery anodes. ACS Nano 4, 1443 (2010).
M. Yoshio, H. Wang, K. Fukuda, T. Umeno, N. Dimov, and Z. Ogumi: Carbon-coated Si as a lithium-ion battery anode material. J. Electrochem. Soc. 149, A1598 (2002).
S.-H. Ng, J. Wang, D. Wexler, K. Konstantinov, Z.-P. Guo, and H.-K. Liu: Highly reversible lithium storage in spheroidal carbon-coated silicon nanocomposites as anodes for lithium-ion batteries. Angew. Chemie Int. Ed. 45, 6896 (2006).
R. Huang, X. Fan, W. Shen, and J. Zhu: Carbon-coated silicon nanowire array films for high-performance lithium-ion battery anodes. Appl. Phys. Lett. 95, 133119 (2009).
H. Kim, X. Huang, Z. Wen, S. Cui, X. Guo, and J. Chen: Novel hybrid Si film/carbon nanofibers as anode materials in lithium-ion batteries. J. Mater. Chem. A 3, 1947 (2015).
Z.W. Pan, Z.R. Dai, L. Xu, S.T. Lee, and Z.L. Wang: Temperature-controlled growth of silicon-based nanostructures by thermal evaporation of SiO powders. J. Phys. Chem. B 105, 2507 (2001).
P.G. Kuzmin, G.A. Shafeev, V.V. Bukin, S.V. Garnov, C. Farcau, R. Carles, B. Warot-Fontrose, V. Guieu, and G. Viau: Silicon nanoparticles produced by femtosecond laser ablation in ethanol: size control, structural characterization, and optical properties. J. Phys. Chem. C 114, 15266 (2010).
Z. Bao, M.R. Weatherspoon, S. Shian, Y. Cai, P.D. Graham, S.M. Allan, G. Ahmad, M.B. Dickerson, B.C. Church, Z. Kang, H.W. Abernathy, C.J. Summers, M. Liu, and K.H. Sandhage: Chemical reduction of threedimensional silica micro-assemblies into microporous silicon replicas. Nature 446(7132), 172 (2007).
Y. Shen, P. Zhao, and Q. Shao: Porous silica and carbon derived materials from rice husk pyrolysis char. Microporous Mesoporous Mater. 188, 46 (2014).
L. Batchelor, A. Loni, L.T. Canham, M. Hasan, and J.L. Coffer: Manufacture of mesoporous silicon from living plants and agricultural waste: an environmentally friendly and scalable process. Silicon 4, 259 (2012).
J. Zhu and D. Deng: Synthesis of curved Si flakes using Mg powder as both the template and reductant and their derivatives for lithium-ion batteries. RSC Adv. 5, 67315 (2015).
P. Lamontagne, G. Soucy, J. Veilleux, F. Quesnel, P. Hovington, W. Zhu, and K. Zaghib: Synthesis of silicon nanowires from carbothermic reduction of silica fume in RF thermal plasma. Phys. status solidi 211, 1610 (2014).
Acknowledgment
The authors would like to thank Dr. Ying Li for supporting chemical treatment facilities and Daebaek, LLC. for silica nano-wires supply for this research. The authors gratefully acknowledge the use of the Texas A&M Materials Characterization Facility (MCF) for SEM. Partial funding for this research was provided by the Meinhard H. Kotzebue ‘14 Professorship endowment in Mechanical Engineering at Texas A&M University.
Author information
Authors and Affiliations
Corresponding author
Supplementary material
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1557/mrc.2017.106
Rights and permissions
About this article

Cite this article
Nam, Y.G., Humood, M., Kim, H. et al. Electrode architecture of carbon-coated silicon nanowires through magnesiothermic reduction for lithium-ion batteries. MRS Communications 7, 867–872 (2017). https://doi.org/10.1557/mrc.2017.106
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/mrc.2017.106