
Abstract
Electronically active block polymers based on π-conjugated macromolecules have been investigated for applications where nanostructured electrodes are of prime import; however, controlling the nanoscale order of these materials has proven challenging. Here, we demonstrate that diblock copolymers that utilize a non-conjugated radical polymer moiety as the electronically active block assemble into ordered thin-film nanostructures. Specifically, the diblock copolymer polydimethylsiloxane-b-poly(2,2,6,6-tetramethylpiperidinyloxy methacrylate) (PDMS–PTMA) was synthesized via atom transfer radical polymerization to generate polymers with readily controlled molecular properties. Importantly, solvent annealing of the PDMS–PTMA thin films led to well-defined nanostructures with domain spacings of the order of ∼30–40 nm.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Block polymers that demonstrate tailored morphologies have been utilized for a range of applications extending from nanolithography[1–3] to membrane separations[4,5] through organic electronic devices[6–8] due to the regular ordering of nanoscale features. Importantly, the thin-film self-assembly behavior of block polymers containing both relatively flexible (coil-like) segments and rigid (rod-like) segments has been investigated heavily from theoretical, computational, and experimental views.[9–12] Recently, Suga et al.[13] demonstrated, for the first time, the formation of thin-film structures in radical containing diblock systems; however, these results did not show well-ordered behavior or long-range order and many questions remain regarding the self-assembly in this class of macromolecules. This is despite the fact that polymers bearing stable radical groups on the side chains of their flexible macromolecular backbones (i.e., radical polymers) are emerging as promising materials in many organic electronic applications (e.g., flexible batteries and polymer-based photovoltaic devices). [14–17] Here, we synthesize well-defined diblock copolymers containing a radical polymer moiety and elucidate the nanostructure of radical polymer-based diblock copolymer thin films. Furthermore, these nanostructured thin films demonstrate the viability of using block polymer motifs as a means by which to create electronically active nano structured radical polymers for myriad organic electronic applications (e.g., nanostructured electrodes in flexible solid-state batteries).[18,19]
The implementation of semiconducting and conducting moieties in block polymer materials is an increasingly studied field because controlling the nanostructure of many organic electronic devices is of prime import.[7,20] However, the use of π-conjugated semiconducting and conducting moieties in block polymer systems usually leads to more complicated microphase separation behavior than that associated with non-conjugated systems.[8] These additional complexities include the fact that the rigid macromolecular backbone of conjugated polymer systems impacts the number of chain conformations available to the block polymer system.[21] Furthermore, in many instances, the conjugated polymer moiety is semicrystalline at room temperature. Therefore, microphase separation between the moieties of the block polymer system due to chemical dissimilarity and the entropy of chain stretching can be dominated by crystallization of the conjugated block.[3] This can be overcome through careful design of the thermal transitions of the semicrystalline moiety of the block polymer,[6,22] but it often leads to block polymer systems that rarely self-assemble into the more oft-observed coil-coil nanostructures (e.g., hexagonally packed cylinders and gyroid structures) that are advantageous for a number of organic electronic applications. [8] As such, the ability to utilize radical polymers with non-conjugated macromolecular backbones provides the important dual effect of: (1) reducing the complexity of thin-film self-assembly, while (2) simultaneously providing a block polymer where at least one moiety is electronically active.
Despite the promise of these synergistic effects, the nanostructured morphologies available to radical polymer-based block polymers have only very recently been discussed. In these important initial studies, diblock copolymers of poly(2,2,6,6-tetramethylpiperidinyloxy methacrylate)-b-polystyrene (PTMA-PS) were synthesized.[23,24] Then, the materials were allowed to self-assemble in solution using electrolytes associated with common energy storage devices. These solution-based self-assembly techniques demonstrated that the chemical dissimilarity between PTMA and other common polymeric species could lead to self-assembled structures in solution. In this effort, we implement atom transfer radical polymerization (ATRP) to readily synthesize well-defined (D ≤ 1.25) polydimethylsiloxane-b-poly(2,2,6,6-tetramethylpiperidinyloxy methacrylate) (PDMS-PTMA) diblock polymers using a PDMS-based macroinitiator. Furthermore, the thin-film nanostructural ordering of diblock copolymers is evaluated systematically across a range of diblock copolymer compositions and molecular weights. Specifically, through proper solvent annealing of the PDMS-PTMA thin films, we establish that a number of microphase separated structures of the diblock copolymer can be observed. This control of the self-assembly of electronically active, radical polymer-based diblock copolymers presents itself as an exciting opening for the rationale design and control of nanostructured organic electronic materials.
Experimental
Materials
All materials were purchased from Sigma-Aldrich and used as received unless indicated otherwise. The macroinitiator, bromine-terminated poly(dimethyl siloxane) (PDMS-Br), with a molecular weight of 10 kg/mol, was purchased from Polymer Source. The monomer, 2,2,6,6-tetramethyl-4-piperidyl methacrylate (TMPM), was purchased from TCI America. The Dowex agent was stirred with ethanol and dichloromethane (DCM) overnight prior to use to remove impurities from the material. The oxidizing agent, meta-chloroperbenzoic acid (mCPBA), was dissolved in ether and washed with water prior to being dried under vacuum overnight.
General methods
Size exclusion chromatography (SEC) data were collected on a Hewlett-Packard 1260 Infinity series equipped with a Hewlett-Packard G1362A refractive index (RI) detector and equipped with three PLgel 5 µm MIXED-C columns. Polystyrene standards (Agilent Easi Cal) of molecular weights ranging from 1 to 200 kg/mol were utilized as calibrants for the SEC data. The mobile phase was tetrahydrofuran (THF) at a temperature of 35 °C flowing at a rate of 1 mL/min. The 1H nuclear magnetic resonance (NMR) spectra were measured on a Bruker DRX500 spectrometer using a ~1% (by weight) polymer solution in deuterated chloroform. The oxidized PDMS–PTMA samples were quenched with hydrazine prior to the NMR spectra collection. Differential scanning calorimetry data were collected on a TA Instruments Q2000 Series calorimeter equipped with a liquid nitrogen cooling unit. Under a helium gas purge, the samples were annealed to 200 °C and held at 200 °C for 10 min. Subsequently, the samples were cooled to [150 °C and held at [150 °C for 10 min. Next, the samples were heated to 200 °C at a heating rate of 10 °C/min, from which the thermal transition temperatures were attained. Thermogravimetric analysis data were collected on a TA SDT Q600 under an air environment. The samples were heated from 25 to 600 °C at a scan rate of 10 °C/min. A Bruker EMX-EPR was utilized for electron paramagnetic resonance (EPR) measurements of a small molecule standard, 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO-OH), and the diblock copolymers a concentration of 2 mg/mL in toluene at room temperature. The attenuated total internal reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) data were collected in 36 scans using a Thermo-Nicolet Nexus FTIR (over the range 800 cm−1 ≤ v ≤ 4500 cm−1), while the ATR-FTIR was under a nitrogen purge. Atomic force microscopy (AFM) imaging was performed using a VEECO Dimension 3100 atomic force microscope operating in tapping mode.
Thin-film separation preparation
The polymer thin films were spin-coated onto glass substrates for AFM imaging. Glass substrates from Delta Technologies were cleaned by sequential sonication in acetone, chloroform, and isopropanol for 10 min each. The substrates were dried with nitrogen and the polymer solutions (20 mg of polymer per 1 mL of chloroform) were spin-coated at a rotational rate of 300 rpm for 3 s prior to ramping to, and holding at, a rotational rate of 600 rpm for 57 s. This procedure led to the formation of ~100 nm thick films, as measured by a KLA Tencor profilometer. The resulting PDMS-PTMA thin films were annealed in chloroform vapors at room temperature for 24 h prior to AFM imaging.
Polymer synthesis
All diblock copolymers were synthesized using an ATRP mechanism where the PDMS-Br served as the macroinitiating agent. The molecular weight of the PTMA unit was tuned by controlling the molar ratio of the monomer present in the reaction solution relative to the concentration of macroinitiation sites. An example PDMS-PTMA diblock copolymer synthesis procedure is detailed below.
PDMS(10)–PTMPM(10)
First, a 150 mL reaction vessel containing a Teflon-coated stir bar was dried overnight. Next, 0.9 g (4 mmol) of TMPM, 4 µL (0.02 mmol) of N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA), 200 µL (0.02 mmol) of the PDMS-Br, and 12 mL of THF were added to the reaction vessel. At this point, the reaction mixture underwent three freeze–pump–thaw cycles and was then placed under an argon blanket. Subsequently, the CuBr catalyst (2.8 mg, 0.02 mmol) was added under this inert atmosphere purge. After the CuBr fully dissolved, the homogeneous solution underwent three additional freeze–pump–thaw cycles. The flask was then refilled with argon and the reaction solution was heated to 65 °C in the closed vessel. After 24 h, the reaction was terminated by cooling the reaction to room temperature and introducing oxygen to the reaction vessel. The solution mixture was stirred at room temperature overnight with a Dowex agent to remove the any residual copper from solution. Next, the solution was injected into a dialysis tube (cut-off molecular weight of 1.2 kg/mol) and stirred at room temperature in THF for 24 h to remove residual monomer from solution. The resulting polymer was dried under reduced pressure overnight.
PDMS(10)–PTMA(10)
The polymer precursor, PDMS(10)–PTMPM(10), was oxidized following a previously established protocol.[25] First, 0.2 g of PDMS(10)-PTMPM(10) was dissolved in 2 mL of DCM at room temperature. A separate solution of mCPBA was made using 0.2 g (1.1 mmol) in 2 mL of DCM. The mCPBA solution was added to the polymer solution slowly while stirring at room temperature and in ambient conditions. The reaction proceeded for 3 h at room temperature. The polymer solution then was washed with an aqueous solution of sodium carbonate (20%, by weight) three times. The polymer solution was precipitated in hexanes, filtered, and the collected solids were dried under reduced pressure overnight.
Results and discussion
Critical to elucidating the thin-film self-assembly of radical-containing block copolymers is synthesizing the PDMS-PTMA diblock copolymers over a wide range of radical-bearing moiety compositions and ensuring that the molecular weight distributions of the diblock copolymers remained relatively narrow.[4,26] Although recent efforts have demonstrated that tuning the dispersity of block polymer samples can be a useful handle in manipulating the observed microstructures of the materials,[27,28] we aimed to eliminate this effect for the purposes of simplicity in our study. To accomplish this objective, we utilized a controlled radical polymerization, atom transfer radical polymerization, which is used commonly for block copolymer synthesis and for the synthesis of PTMA homopolymers.[23,29]
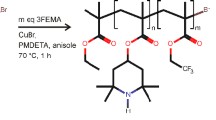
In this work, the first block (i.e., PDMS-Br) of known molecular weight was utilized as the macroinitiator from which the PTMPM block was grown (Fig. 1). Here PDMS was used as the insulating moiety due to the high chain mobility of PDMS at and above room temperature. This helped to facilitate the nanostructural ordering observed in the diblock copolymer thin-film samples upon solvent annealing (vide infra). Following two purification steps, the PDMS–PTMPM diblock copolymers were analyzed using 1H NMR to determine their purity as well as the molecular weight of the PTMPM moieties (Fig. S1a). Because of the controlled nature associated with the ATRP mechanism, the molecular weight of the PTMPM segment could be tuned in a rather straightforward manner (i.e., through control of the relative ratios of the polymerization reagents) and, the molecular weights of the PTMPM blocks were determined readily through integration of the proton resonances associated with the methyl protons on the silicon of the PMDS-Br backbone and the proton on the PTMPM pendant ring (Fig. 2a).

The ATRP-mediated route employed for the synthesis of PDMS–PTMA using PDMS-Br as the macroinitiator, PMDETA as the ATRP ligand, CuBr as the ATRP catalyst, and TMPM as the monomer for the second block. The resulting PDMS–PTMPM was oxidized with mCPBA to form the radical copolymer PDMS-PTMA.

(a) A representative 1H NMR spectrum for the purified PDMS(10)–PTMPM(10) diblock copolymer dissolved in deuterated chloroform (CDCl3). The integration of the peaks associated with the resonances labeled A and B allowed for the number-average molecular weight of the PTMPM moiety to be determined in a straightforward manner. (b) SEC traces for the PDMS-Br homopolymer and representative PDMS–PTMA diblock copolymers over the entire range of the diblock copolymers synthesized. All SEC experiments were performed with a mobile phase of THF flowing at a rate of 1 mL/min and at temperature of 35 °C. Dispersity values were calculated relative to PS standards.
In particular, the ATRP scheme employed was capable of synthesizing PTMA moieties with molecular weight values that ranged from 0 to 7 kg/mol in a straightforward fashion. Next, utilizing a previously established method,[30] the parent polymers (i.e., PDMS–PTMPM) were converted to the radical-bearing functionality using mCPBA as the oxidizing agent to produce the PDMS–PTMA diblock copolymers. We note that the oxidation procedure did not affect the PDMS block of the diblock copolymer, as was the case in previous reports where polystyrene (PS) was utilized as the second block of a PTMA-based diblock copolymer.[23] After quenching the radical sites with hydrazine, the H NMR spectra of the PDMS–PTMA diblock copolymers were acquired (Fig. S1b), and these data confirmed the integrity of the PDMS and PTMA moieties. Moreover, the SEC traces of the PDMS–PTMA samples demonstrate a clear shift in elution time corresponding well to the increasing molecular size in the diblock system (Fig. 2b). Additionally, the dispersity values of all the PDMS–PTMA samples synthesized remained relatively low (i.e., D < 1.25), confirming that the polymerization occurred in a controlled manner (Table I). We do note that, for the higher molecular weight PTMA blocks [e.g., PDMS(10)–PTMA(36) and PDMS(10)–PTMA(71)], a slight shoulder at lower elution volumes is observed; however, the dispersity values of these samples also remained at values of D ≤ 1.25.
The chemical functionality of the PDMS–PTMPM and PDMS–PTMA diblock copolymers in the solid state was confirmed through ATR-FTIR, as the expected signals from the key functional groups of the block polymers were readily visible (Fig. S2). Furthermore, the specific presence of the radical moieties pendant to each of the repeat units were monitored through EPR spectroscopy, as has been shown previously for numerous electronically active radical polymer systems.[20,30,31] The absorption of the nitroxide radical is well established and occurs at a magnetic field of 3358 G,[30] which corresponds very well with the PDMS–PTMA absorption (Fig. 3). To quantify the degree to which the PTMPM moiety was converted to the PTMA moiety (i.e., the fraction of repeat units on the PTMA chain that contained a stable radical group) the EPR absorption spectra were normalized to the small molecule TEMPO-OH absorption spectrum (Fig. S3). This is because the TEMPO-OH signal is similar to the nitroxide functionality associated with PTMA, and it is known that each TEMPO-OH molecule contains a single radical site. In this way, a direct comparison could be made. As shown in Table I, all of the samples showed a reasonable (i.e., ≥35%) fraction of radical groups. Furthermore, the fraction of radical groups present was relatively consistent between the PDMS–PTMA diblock copolymers. As such, we are confident that the chemical dissimilarity between the two segments of the PDMS–PTMA diblock copolymers that self-assemble (described below) are, to first order, the same.

(a) EPR spectra of a range of PDMS–PTMA diblock copolymers in toluene at a concentration of 2 mg/mL. These data demonstrate the higher absorption (i.e., higher radical density) for PTMA moieties with weight fractions of PTMA present in the PDMS–PTMA diblock copolymers. Additionally, the PDMS–PTMA diblock copolymers with larger molecular weights of PTMA demonstrate hyperfine splitting, suggesting less radical-to-radical interactions. As an example of this, (b) isolates the EPR spectra for PDMS(10)–PTMA(71) and PDMS(10)–PTMA(19) and PMDS(10)-PTMA(71).
Interestingly, as the molecular weight of the PTMA block increases beyond 20 kg/mol (i.e., wPTMA ≥ 0.70), it is evident that splitting of the absorption peaks occurs (Fig. 3a). This phenomenon (i.e., hyperfine splitting) is indicative of reduced radical-to-radical interactions in the EPR solution (Fig. 3b).[31] Importantly, the hyperfine splitting suggests that, as the size of the PTMA block increases, the polymer chains adopt a different conformation in solution. In this conformation, the radical sites are necessarily farther removed in space despite the fact that a larger percentage of the diblock copolymer contains radical sites.

Summary of PDMS–PTMA molecular and thermal properties.
While the solution-state behavior of PDMS–PTMA is notable and worthy of further study, to be of high utility in organic electronic applications (e.g., solid-state batteries), the ability of these PDMS–PTMA materials to generate well-defined nanostructures as thin films is key. Although this has been an important point of research for optoelectronically active block polymers based on π-conjugated macromolecules, only a few select efforts have been able to show distinct assemble of these materials that is unique from what is observed due to the crystallization of the homopolymer conjugated polymer. By utilizing non-conjugated, amorphous radical polymers as the electronically active moiety of the block copolymer, this work circumvents this common roadblock. As expected for coil-coil diblock copolymer thin films, nanoscale features were observed over a large range of polymer molecular weights when the PDMS-PTMA diblock copolymers were cast as thin films. Owing to the rather high glass transition temperature of PTMA (Table I) and the unstable thermal nature of the radical group associated with the nitroxide group (i.e., degradation of the polymer beings to occur at T ~210 °C, Fig. S4), thermal annealing of these materials is very difficult. Instead, solvent annealing in chloroform vapor was implemented to provide mobility to both the PDMS and PTMA moieties of the diblock copolymer. For diblock copolymers containing a higher weight fraction of the relatively mobile PDMS moiety, the exposure to chloroform vapors led to relatively well defined, nanostructured PDMS-PTMA (Fig. 4), as imaged with AFM. The domain sizes observed were of the order of 30–40 nm for all samples that showed clear nanostructural ordering (i.e., those with higher PDMS content). However, as the weight fraction of the PTMA block is increased, the ability to form ordered thin-film nanostructures is decreased through the use of solvent annealing (Fig. 4c). This seems to be associated with the limited mobility of the PTMA block, and it can be attributed to its high glass transition (i.e., >150 °C). This high thermal transition is well-known in PTMA, and it occurs due to the relatively bulky and polar 2,2,6,6-tetramethylpiperidinyloxy-4-yl side chain of the macromolecule.[25] That is, because of the similar radical conversion in almost all of the PDMS–PTMA samples, this lack of ordering at higher PTMA molecular weights does not seem to be due to a change in the chemical nature of the radical polymer moiety. It is critical to note, however, that the self-assembly of this macromolecular system is highly dependent on the radical functionality being present in the PDMS–PTMA system. That is, no evidence of nanostructure is observed for thin films of the PDMS–PTMPM diblock systems (Fig. S5).

AFM phase images of (a) PDMS(10)–PTMA(10), (b) PDMS(10)–PTMA(36), and (c) PDMS(10)–PTMA(51) thin films after annealing with chloroform vapors for 24 h at room temperature. The PDMS(10)–PTMA(10) and the PDMS(10)-PTMA(36) systems showed ordered microstructures. Samples with large weight fractions of PTMA did not order in the thin film after exposure to the solvent-annealing procedure.
Therefore, great care must be taken when designing the composition, molecular weight, and functionality of PTMA-containing diblock copolymers and the specific annealing conditions used must be well-tailored if the thin-film assembly of the materials is a prime component of application success. Fortunately, these design parameters can be implemented in a straightforward manner due to the large number of controlled synthetic techniques available to radical polymer-based block polymers relative to conjugated polymer-based block polymers.
Conclusions
In summary, radical-containing diblock copolymers were synthesized via an ATRP mechanism to yield electronically active macromolecules with controlled molecular weights, narrow molecular weight distributions, and a variety of block polymer compositions. Characterization of the paramagnetic centers in the PTMA moieties of the diblock copolymers indicated that little overlap between the PTMA units was present for PDMS–PTMA samples that contained a relatively larger weight fraction of PTMA. Furthermore, when cast into thin films these diblock copolymers self-assemble into ordered nanostructures, when there is sufficient mobility of the diblock copolymers. Here, this mobility was imparted through solvent annealing using chloroform vapors; however, for PDMS–PTMA samples that contained larger weight fractions of the radical polymer moiety, the well-ordered structures were difficult to observe. The lower molecular weight PTMA–PDMS samples are the first initial demonstration of continuous thin-film ordering on the nanoscale from radical polymer-containing block polymers. Therefore, these data provide a methodology by which to create a number of functional electronic materials. In particular, it provides a solid foundation by which to explore systems where the second block of a diblock copolymer has a complementary functionality (e.g., the utilization of polymers capable of transporting lithium ions efficiently) to that of the electronically active radical polymer component. Therefore, these platform radical polymer-based materials, with easily controlled molecular and nanostructural properties, could play a significant role in organic electronic applications requiring control of electrode structure at the nanoscale level.
References
J.G. Kennemur, L. Yao, F.S. Bates, and M.A. Hillmyer: Sub-5 nm domains in ordered poly(cyclohexylethylene)-block-poly(methyl methacrylate) block polymers for lithography. Macromolecules 47, 1411 (2014).
J. Xu, T.P. Russell, B.M. Ockob, and A. Checco: Block copolymer selfassembly in chemically patterned squares. Soft Matter 7, 3915 (2011).
M. Luo and T.H. Epps III: Directed block copolymer thin film selfassembly: emerging trends in nanopattern fabrication. Macromolecules 46, 7567 (2013).
Y. Zhang, J.L. Sargent, B.W. Boudouris, and W.A. Phillip: Nanoporous membranes generated from self-assembled block polymer precursors: Quo Vadis? J. Appl. Polym. Sci. 132, 41683 (2015).
E.A. Jackson and M.A. Hillmyer: Nanoporous membranes derived from block copolymers: from drug delivery to water filtration. ACS Nano 4, 3548 (2010).
B.W. Boudouris, V. Ho, L.H. Jimison, M.F. Toney, A. Salleo, and R.A. Segalman: Real-time observation of poly(3-alkylthiophene) crystallization and correlation with transient optoelectronic properties. Macromolecules 44, 6653 (2011).
C. Renaud, S.-J. Mougnier, E. Pavlopoulou, C. Brochon, G. Fleury, D. Deribew, G. Portale, E. Cloutet, S. Chambon, L. Vignau, and G. Hadziioannou: Block copolymer as a nanostructuring agent for high efficiency and annealing-free bulk heterojunction organic solar cells. Adv. Mater. 24, 2196 (2012).
B.D. Olsen and R.A. Segalman: Self-assembly of rod–coil block copolymers. Mater. Sci. Eng. 62, 37 (2008).
B.D. Olsen, X. Li, J. Wang, and R.A. Segalman: Thin film structure of symmetric rod-coil block copolymers. Macromolecules 40, 3287 (2007).
M. Shah and V. Ganesan: Correlations between morphologies and photovoltaic properties of rod-coil block copolymers. Macromolecules 43, 543 (2010).
C. Sinturel, D. Grosso, M. Boudot, H. Amenitsch, M.A. Hillmyer, A. Pineau, and M. Vayer: Structural transitions in asymmetric poly (styrene)-block-poly(lactide) thin films induced by solvent vapor exposure. ACS Appl. Mater. Interfaces 6, 12146 (2014).
W.A. Phillip, M.A. Hillmyer, and E.L. Cussler: Cylinder orientation mechanism in block copolymer thin films upon solvent evaporation. Macromolecules 43, 7763 (2010).
T. Suga, M. Sakata, K. Aoki, and H. Nishide: Synthesis of pendant radicaland ion-containing block copolymers via ring-opening metathesis polymerization for organic resistive memory. ACS Macro Lett. 3, 703 (2014).
L. Rostro, L. Galicia, and B.W. Boudouris: Suppressing the environmental dependence of the open-circuit voltage in inverted polymer solar cells through a radical polymer anodic modifier. J. Polym. Sci. B, Pol. Phys. 53, 311 (2015).
K. Oyaizu and H. Nishide: Radical polymers for organic electronic devices: a radical departure from conjugated polymers? Adv. Mater. 21, 2339 (2009).
T. Janoschka, M.D. Hager, and U.S. Schubert: Powering up the future: radical polymers for battery applications. Adv. Mater. 24, 6397 (2012).
E.P. Tomlinson, M.E. Hay, and B.W. Boudouris: Radical polymers and their application to organic electronic devices. Macromolecules 47, 6145 (2014).
W.-S. Young, P.J. Brigandi, and T.H. Epps III: Crystallization-induced lamellar-to-lamellar thermal transition in salt-containing block copolymer electrolytes. Macromolecules 41, 6276 (2008).
S.N. Patel, A.E. Javier, and N.P. Balsara: Electrochemically oxidized electronic and ionic conducting nanostructured block copolymers for lithium battery electrodes. ACS Nano 7, 6056 (2013).
S.N. Patel, A.E. Javier, K.M. Beers, J.A. Pople, V. Ho, R.A. Segalman, and N.P. Balsara: Morphology and thermodynamic properties of a copolymer with an electronically conducting block: poly(3-ethylhexylthiophene)- block-poly(ethylene oxide). Nano Lett. 12, 4901 (2012).
C. Singht, M. Goulian, A.J. Lid, and G.H. Fredrickson: Phase behavior of semiflexible diblock copolymers. Macromolecules 27, 2974 (1994).
V. Ho, B.W. Boudouris, B.L. McCulloch, C.G. Shuttle, M. Burkhardt, M.L. Chabinyc, and R.A. Segalman: Poly(3-alkylthiophene) diblock copolymers with ordered microstructures and continuous semiconducting pathways. J. Am. Chem. Soc. 133, 9270 (2011).
G. Hauffman, J. Rolland, J.-P. Bourgeois, A. Vlad, and J.-F. Gohy: Synthesis of nitroxide-containing block copolymers for the formation of organic cathodes. J. Polym. Sci. Pol. Chem. 51, 101 (2013).
G. Hauffman, Q. Maguin, J.-P. Bourgeois, A. Vlad, and J.-F. Gohy: Micellar cathodes from self-assembled nitroxide-containing block copolymers in battery electrolytes. Macromol. Rapid Commun. 35, 228 (2014).
L. Rostro, A.G. Baradwaj, and B.W. Boudouris: Controlled radical polymerization and quantification of solid state electrical conductivities of macromolecules bearing pendant stable radical groups. ACS Appl. Mater. Interfaces 5, 9896 (2013).
F.S. Bates and G.H. Fredrickson: Block copolymer thermodynamics: theory and experiment. Annu. Rev. Phys. Chem. 41, 525 (1990).
J.M. Widin, M. Kim, A.K. Schmitt, E. Han, P. Gopalan, and M.K. Mahanthappa: Bulk and thin film morphological behavior of broad dispersity poly(styrene-b-methyl methacrylate) diblock copolymers. Macromolecules 46, 4472 (2013).
A.L. Schmitt, M.H. Repollet-Pedrosa, and M.K. Mahanthappa: Polydispersity-driven block copolymer amphiphile self-assembly into prolate-spheroid micelles. ACS Macro Lett. 1, 300 (2012).
K. Matyjaszewski: Atom transfer radical polymerization (ATRP): current status and future perspectives. Macromolecules 45, 4015 (2012).
L. Rostro, S.H. Wong, and B. Boudouris: Solid state electrical conductivity of radical polymers as a function of pendant group oxidation state. Macromolecules 47, 3713 (2014).
D.C. Bobela, B.K. Hughes, W.A. Braunecker, T.W. Kemper, R.E. Larsen, and T. Gennett: Close packing of nitroxide radicals in stable organic radical polymeric materials. J. Phys. Chem. Lett. 6, 1414 (2015).
Acknowledgments
We gratefully acknowledge financial support from the Air Force Office of Scientific Research through the Young Investigator Program (AFOSR YIP, Grant number FA9550-12-1-0243, Program Manager: Dr. Charles Lee). L. R. and J. S. L. appreciatively acknowledge the National Science Foundation forsupport through the Graduate Research Fellowship Program (Grant number DGE-1333468). A. R. M. thanks the National Science Foundation for partial support of his work through the Nanotechnology Undergraduate Education (NUE) in Engineering Program (Award number 1242171, Program Manager: Dr. Mary Poats). Further support of the work of A. R. M. was generously provided by the Intel Corporation and the Semiconductor Research Corporation (SRC) Education Alliance. We thank Gamini Mendis for assistance with the differential scanning calorimetry experiments.
Author information
Authors and Affiliations
Corresponding author
Supplementary materials
Supplementary materials
For supplementary material for this article, please visit http://dx.doi.org/10.1557/mrc.2015.27
Rights and permissions
About this article

Cite this article
Rostro, L., Baradwaj, A.G., Muller, A.R. et al. Synthesis and thin-film self-assembly of radical-containing diblock copolymers. MRS Communications 5, 257–263 (2015). https://doi.org/10.1557/mrc.2015.27
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/mrc.2015.27