
Abstract
A novel g-C3N4 nanoparticle@porous g-C3N4 (CNNP@PCN) composite has been successfully fabricated by loading g-C3N4 nanoparticles on the porous g-C3N4 matrix via a simply electrostatic self-assembly method. The composition, morphological structure, optical property, and photocatalytic performance of the composite were evaluated by various measurements, including XRD, SEM, TEM, Zeta potential, DRS, PL, FTIR, and XPS. The results prove that the nanolization of g-C3N4 leads to an apparent blueshift of the absorption edge, and the energy band gap is increased from 2.84 eV of porous g-C3N4 to 3.40 eV of g-C3N4 nanoparticle (Fig. 6). Moreover, the valence band position of the g-C3N4 nanoparticle is about 0.7 eV lower than that of porous g-C3N4. Therefore, the photo-generated holes and electrons in porous g-C3N4 can transfer to the conduction band of g-C3N4 nanoparticle, thereby obtaining higher separation efficiency of photo-generated carriers as well as longer carrier lifetime. Under visible-light irradiation, 6CNNP@PCN exhibits the highest photocatalytic performance (Fig. 8) on MB, which is approximately 3.4 times as that of bulk g-C3N4.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In 1972, Fujishima and Honda firstly reported on Nature that rutile single-crystal TiO2 can be used as an efficient photocatalyst. Since then, photocatalytic materials have aroused great interest owing to its features of renewability, stability, and safety [1, 2, 3]. Developing high-performance semiconductor photocatalysts has been considered to be an effective way to solve the problem of energy shortage and dye pollution. Although photocatalysis research has made much progress in recent decades, pursuing higher degradation efficiency of photocatalysts and better availability of visible light are still challenging issues and have always been the significant research emphasis in the photocatalytic field [4, 5, 6, 7, 8, 9, 10].
Nowadays, it is well-known that graphitic carbon nitride [11, 12, 13] (g-C3N4) has attracted great interest because of its two-dimension and tunable electronic structure as well as an appropriate band gap (2.7 eV) and excellent stability [14, 15, 16, 17, 18]. Nevertheless, bulk g-C3N4 synthesized through a traditional polycondensation process still exists a series of defects, such as the rapid recombination of photo-generated carriers and low absorption of visible light [19, 20]. Consequently, numerous experiments have been investigated to enhance the photocatalytic performance of g-C3N4, like microstructure modification [21], metal element doping [22], and construction of heterojunction [23, 24, 25, 26, 27, 28, 29]. At present, researchers have built a variety of g-C3N4-based composite systems, such as SnO2/g-C3N4 [30], SnS2/g-C3N4 [31], ZnWO4/g-C3N4 [32], BiPO4/g-C3N4 [33], and Ag3PO4/g-C3N4 [34, 35]. Zhang et al. [36] prepared g-C3N4/BiOI composite via the solid-phase calcination method with the enhanced photocatalytic performance for MC-LR degradation. Liu et al. [37] synthesized Bi2O3/g-C3N4 composite by means of a facile one-step fabrication with improved photocatalytic activity. Yan et al. [38] obtained a ternary Ag3VO4/C3N4/reduced TiO2 composite through a hydrothermal and depositional process, revealing the excellent photodegradation activity. Lu et al. [39, 40] designed Ni3B/Ni(OH)2 over g-C3N4 nanosheet and MPx (M = Fe, Co, or Ni) over g-C3N4/MoS2 layered heterojunctions for H2 evolution. Shen et al. [41] modified Co1.4Ni0.6P as dual cocatalysts over metallic carbon black/g-C3N4 nanosheet for photocatalytic H2 evolution. However, the compounding of different kinds of materials often has an interface mismatch problem due to the difference in the crystal structure, leading to poor carrier migration [42, 43, 44].
In this work, the g-C3N4 nanoparticles (CNNP) were successfully synthesized, which exhibited a different energy band structure from the traditional bulk g-C3N4 (CN). This feature made it possible to combine g-C3N4 nanoparticles with bulk g-C3N4 to realize the migration and separation of photo-generated carriers. More importantly, almost the same crystal structure could avoid the mismatch of the composite interface. Herein, a simply electrostatic self-assembly approach was proposed to load g-C3N4 nanoparticles on porous bulk g-C3N4 (PCN), forming the surface-dispersive-type semiconductor junctions with high photo-generated carrier separation efficiency [45]. Compared with the pure g-C3N4, the photocatalytic activities of the composite enhanced significantly. Moreover, the g-C3N4 nanoparticles contrasted with the bulk g-C3N4 may minimize the interface mismatch with other photocatalyst, and thus, this work also provided a facile approach to construct g-C3N4-decorated photocatalytic composite materials with surface-dispersive-type semiconductor junctions.
Results And Discussion
Structures and morphology
The X-ray diffraction (XRD) patterns of CN, PCN, and CNNP@PCN composites are described in Fig. 1(a). It can be clearly seen that except for CNNP, all samples exhibit two diffraction peaks at 13.2° and 27.4°, belonging to the characteristic diffraction peaks of g-C3N4. The former belongs to the (100) crystal plane of g-C3N4, corresponding to the periodic arrangement of the heptazine structure, which is the basic structural unit in the g-C3N4 layer [9]. The latter belongs to the typical interlayer stacking peaks of aromatic ring series, corresponding to the (002) crystal plane of g-C3N4. For CNNP, the diffraction peak at 13.2° almost disappears due to the geometric restriction effect of nanoparticles. The diffraction peak of CNNP at 27.4° shifts to a large angle compared with CN, and the PCN also has the same phenomenon because of the more intense layer structure [46]. To further investigate the phase structure, the Fourier-transform infrared spectroscopy (FTIR) spectra of CN, PCN, and CNNP@PCN composites are depicted in Fig. 1(b). It can be analyzed that all samples show characteristic peaks [47] of g-C3N4 at 809 and 1200–1600 cm−1 due to the stretching modes of C–N heterocycles, which can prove the integrity of the structure. The results of FTIR and XRD patterns suggest that the main structure of g-C3N4 has not been destroyed during the preparation process.

(a) XRD patterns and (b) FTIR spectra of CN, PCN, and CNNP@PCN composites.
X-ray photoelectron spectroscopy (XPS) analysis was measured to investigate the elemental compositions and chemical status of CN, PCN, and CNNP@PCN composites. The survey XPS spectrums of CN, PCN, and CNNP@PCN composites obviously exhibit the presence of C and N elements. As displayed in Fig. 2(b), the C 1s spectrum can be fitted to two peaks at 284.6 and 287.9 eV. The peak at 287.9 eV is attributed to the sp2-bonded carbon in the CN network and the peak at 284.6 eV is ascribed to sp2 C–C bonds [11, 48]. The peak of N 1s spectra located at 398.1 eV is attributed to the sp2-bonded nitrogen in triazine rings (C–N=C) [37]. Notably, both the peaks of C 1s and N 1s for the 6CNNP@PCN shift toward the higher binding energies, indicating the probably higher electron transfer efficiency and interfacial interaction between g-C3N4 nanoparticles and porous g-C3N4 in the composite.

(a) XPS survey spectra and the corresponding high-resolution XPS spectra of (b) C 1s and (c) N 1s of CN, PCN, and 6CNNP@PCN.
The surface morphology and structure of CN, PCN, CNNP, and CNNP@PCN composites were measured by scanning electron miscroscopy (SEM) in Fig. 3. As depicted in Figs. 3(a)–3(c), compared with bulk CN, PCN has the interconnected channels and porous structure with a large area exposed and smooth surface consisting of layered g-C3N4 [49, 50], which is conducive to the loading of g-C3N4 nanoparticles. In addition, the size of PCN was approximately 3–5 μm, while the size of CNNP was 30–50 nm, making it possible for the designed heterojunction microstructure. Therefore, it can be successfully observed from Fig. 3(e) that g-C3N4 nanoparticles are homogeneously loaded on the surface of porous g-C3N4. Meanwhile, with the loading content of g-C3N4 nanoparticles increasing from 1% to 10% [Figs. 3(e)–3(g)], it can be seen that the load rate of g-C3N4 nanoparticles on porous g-C3N4 enhances as well. However, excessive g-C3N4 nanoparticles not only occupies the surface position of porous g-C3N4 but also forms aggregation to block the porous structure of porous g-C3N4.

SEM images and corresponding high-magnification SEM images (insets) of (a) CN, (b) PCN, (c) CNNP, (d) 1CNNP@PCN, (e) 3CNNP@PCN, (f) 6CNNP@PCN, and (g) 10CNNP@PCN.
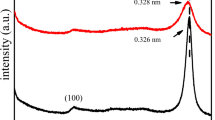
To further investigate the microstructure and morphology details of 6CNNP@PCN, transmission electron microscope (TEM) observation was carried out. As depicted in Fig. 4, porous g-C3N4 has a large area exposed and smooth surface. Some nanoparticles are dispersedly loaded on the porous g-C3N4, and its calculated d-value in the HRTEM image is 0.325 nm, corresponding to the (0 0 2) facet of g-C3N4. It indicates that g-C3N4 nanoparticles and porous g-C3N4 have formed a close interface, avoiding the mismatch of the composite interface. As a matter of fact, the fabrication of CNNP@PCN is mainly based on the electrostatic self-assembly process. As shown in Supplementary Fig. S1, the surface charge of CNNP is positive in deionized water and its Zeta potential is 21.5 mV, while the Zeta potential of PCN is −9.97 mV. Therefore, g-C3N4 nanoparticles can be easily assembled on the porous g-C3N4 surface under electrostatic action. Additionally, the N2 adsorption–desorption isotherm of CN, PCN, and 6CNNP@PCN composites are depicted in Supplementary Fig. S2. The typical type IV isotherm of as-prepared samples featuring a distinct H1 hysteresis loop reveals the presence of micropores and mesopores [12]. The Brunauer–Emmett–Teller (BET) specific surface area of PCN and 6CNNP@PCN is calculated to be 34.81 and 36.30 m2/g, which is larger than that of CN (11.86 m2/g). It is worth noting that the specific surface area of PCN and 6CNNP@PCN is similar, implying that the load of g-C3N4 nanoparticles on the porous g-C3N4 has little impact on the improvement of the specific surface area.

(a and b) TEM images with different magnifications and (c) HRTEM image of 6CNNP@PCN.
Optical properties
The UV–Vis diffuse reflectance spectra (DRS) of CN, PCN, CNNP, and CNNP@PCN composites were measured to obtain the optical absorption properties, as shown in Fig. 5. The CN holds an absorption edge at 450 nm, which is accordant with that reported in the literature [51]. Compared with CN, the absorption edge of PCN clearly blueshifts to around 430 nm, which is related to its porous structure [52]. Moreover, the absorption edge of CNNP contrasted with PCN has a further blueshift due to the nanocrystalline of g-C3N4. The band gap can be directly calculated with the following equation [53]:
where α, ν, Eg, and A are the absorption coefficient, light frequency, band gap energy, and the proportionality constant, respectively. The value of n represents the type of transition (for direct transition, n = 1). Based on the UV–Vis DRS, the plots of (αhʋ)2 versus the photon energy for g-C3N4 nanoparticles were calculated as the value of n is 1 for pure g-C3N4. As demonstrated in Fig. 5(a), the Eg of CN, PCN, and CNNP can be calculated as 2.76, 2.84, and 3.40 eV. While the Eg of CNNP@PCN composites is about 2.85, 2.86, 2.88, and 2.89 eV for 1CNNP@PCN, 3CNNP@PCN, 6CNNP@PCN, and 10CNNP@PCN composites, respectively [Fig. 5(b)]. The results imply that the load of g-C3N4 nanoparticles make an influence on the valence band (VB) position and the conduction band (CB) position of the CNNP@PCN composites, which probably affects the photocatalytic activity.

(a) UV–Vis DRS spectra of CN, PCN, and CNNP@PCN composites; (b) band gap analysis of CNNP, CN, and PCN; and (c) band gap analysis of CNNP@PCN composites.
To further investigate the VB position of PCN and CNNP, XPS measurements were performed. Figure 6 indicates that the VB position of CNNP and PCN are 2.51 and 1.81 eV, respectively. Combining the band gap width calculated from Fig. 5(a), the CB position of CNNP and PCN can be estimated to be about −0.89 and −1.03 eV, respectively.

VB XPS spectra of (a) PCN and (b) CNNP.
The photoluminescence (PL) spectra of CN, PCN, and CNNP@PCN composites are shown in Fig. 7. As depicted in Fig. 7(a), the emission peak of PCN is located at about 460 nm, which exhibits a slightly blueshift compared with that of CN (470 nm). The emission peak strength of the CNNP@PCN composite at 460 nm is less than those of PCN, which indicate that the loading of g-C3N4 nanoparticles on porous g-C3N4 promotes the migration and separation of photo-generated holes and electrons. In reality, this is related to the structure of PCN. Compared with CN, PCN consisting of nanoscale lamellar and porous structures have larger band gap width, which means that the position of the CB and VB shifts negatively and positively, resulting in blueshift and the increase of PL intensity [54]. The 6CNNP@PCN shows the highest photo-generated carrier separation efficiency. Moreover, it is worth noting that 6CNNP@PCN has higher separation efficiency of photo-generated carriers than 6CNNP@CN [Fig. 7(b)]. This is mainly because the PCN has a larger surface area as well as more active sites, resulting in more uniformly and effectively loading of CNNP.

PL spectra of (a) CN, PCN, and CNNP@PCN composites; and (b) 6CNNP@CN and 6CNNP@PCN.
Moreover, time-resolved fluorescence spectroscopy was measured to further study the separation ability of photo-generated carriers. The fluorescence decay curve is fitted by three exponential decay kinetic equations, and the fluorescence lifetime and proportion of each part are obtained (Supplementary Fig. S3 and Table S1). The average fluorescence lifetime of 6CNNP@PCN is 8.27 ns, which is higher than that of CN (5.64 ns), PCN (4.39 ns), and 6CNNP@CN (7.81 ns). This result further confirms that the loading of g-C3N4 nanoparticles can effectively suppress the recombination of photo-generated carriers in the CNNP@PCN as well as CNNP@CN composites.
Photocatalytic performance and possible mechanism
The photocatalytic performance of CN, PCN, and CNNP@PCN composites with different g-C3N4 nanoparticles contents were analyzed by the degradation of MB (methylene blue, 10 mg/L) in aqueous solution under visible light (λ > 420 nm). As depicted in Fig. 8(a), there is a blank test without any catalysts to be performed as a reference. The PCN shows higher photocatalytic activity than CN due to its inter-connected channels and porous structure. Meanwhile, the composite reveals better adsorption ability (Supplementary Fig. S4) than that of CN and PCN, which is beneficial to increasing photocatalytic reaction efficiency. As expected, the composite photocatalyst exhibits higher degradation efficiency than the corresponding pure one, and the 6CNNP@PCN has the highest photocatalytic performance. Consequently, the photocatalytic decomposition kinetics of CN, PCN, and CNNP@PCN composites is fitted by a pseudo-first-order model, which is calculated by the following equation:
where C0, C, and C1 correspond to the origin concentration, the concentration at time t of MB, and the concentration after adsorption, respectively, and k is the apparent first-order rate constant. As depicted in Fig. 8(b), 6CNNP@PCN displays the best and strongest photocatalytic degradation rate of 0.906 h−1 which is 3.4, 1.6, and 1.8 times higher than that of CN (0.265 h−1), PCN (0.551 h−1), and 6CNNP@PCN (0.499 h−1). There is the similar porous g-C3N4/BiOI system reported in the literature [17]. Compared with the CNNP@PCN composite, it is found that the BiOI nanosheet cannot be loaded uniformly on the porous g-C3N4, and the contact area between the two semiconductors is also reduced. Therefore, the CNNP@PCN composite exhibits relatively higher photocatalytic activity than that of g-C3N4/BiOI under the similar photocatalytic reaction conditions. Moreover, the influence of pH of the solution on the photocatalytic degradation process was carried out in Supplementary Fig. S5. The initial pH of MB solution was set in the range of pH 4 to 10 by adjusting with the HCl and NaOH solutions [55, 56]. It can be observed that the photocatalytic activity greatly reduces in the acidic solution, while the photocatalytic activity obviously enhances in the alkaline solution. The phenomena might be explained [57] by the fact that the surface of CNNP@PCN composite is protonated and exhibits a positive charge in the pH range of 4–6, which reduces the adsorption of the positively charged MB by electrostatic repulsion. In the pH range of 8–10, the surface of CNNP@PCN composite becomes negative and the electrostatic attraction could occur, leading to strong adsorption and promoting photocatalytic degradation. In addition, the O2 in MB solution under the alkaline condition captured a large amount of negative charges [58], which may generate numerous \( \cdot {{\text{O}}_{\text{2}}}^ - \), thus enhancing the efficiency of degradation.

(a) Photocatalytic degradation of MB; (b) the corresponding kinetics; DMPO spin-trapping EPR spectra under visible-light irradiation (λ > 420 nm) for 180 s in (c) aqueous solutions for \( \cdot {{\text{O}}_{\text{2}}}^ - \) and (d) methanol solutions for ⋅OH.
Moreover, electron paramagnetic resonance (EPR) was applied to reveal the active species in the photocatalytic reaction process [59, 60]. As displayed in Figs. 8(c) and 8(d), four typical peaks of DMPO-\( \cdot {{\text{O}}_{\text{2}}}^ - \) can be observed under visible-light irradiation, while no signals can be detected in dark. However, the characteristic peaks of DMPO-⋅OH cannot be figured out due to the location of VB when the light turns on. The EPR results prove that the \( \cdot {{\text{O}}_{\text{2}}}^ - \) can be generated on the CB of CNNP@PCN and plays an important role in the photocatalytic system, while ⋅OH cannot be yielded.
Therefore, the degradation mechanism of CNNP@PCN can be explained as follows (Fig. 9): When exposed to visible light, PCN is stimulated to produce photo-generated carries, then because the CB position of PCN is −1.03 eV, which is more negative than that of CNNP −0.89 eV, so the photo-generated electrons located on the PCN CB can be transferred to CNNP and react with the oxygen adsorbed on the surface to produce active \( \cdot {{\text{O}}_{\text{2}}}^ - \), and photo-generated holes will remain at the VB level of PCN and directly participate in the degradation of dyes. Furthermore, the composite system has higher separation efficiency of photo-generated electrons than PCN possibly due to the high migration of photo-generated electrons from PCN by CNNP.

Photo-generated electron–hole pair separation and transfer mechanism in the CNNP@PCN composite.
Conclusion
In summary, a semiconductor junction between porous g-C3N4 and g-C3N4 nanoparticles has been successfully constructed by a simply electrostatic self-assembly method. With this specific microstructure, CNNP@PCN exhibits greatly enhanced photocatalytic performance in MB degradation in contrast with those of CN and PCN under visible-light irradiation. The reasons for the increased photocatalytic performance of the composites may be proposed from more efficient separation and migration of photo-generated carriers within the semiconductor junction. Subsequently, in contrast with bulk g-C3N4, the smaller-size g-C3N4 nanoparticles may diminish the interface mismatch with other promising semiconductors, so it's easy to load the g-C3N4 nanoparticles on series of other photocatalysts with a matched energy band structure as well as offering a new method to structure photocatalytic materials decorated with g-C3N4.
Experimental Section
Materials
Melamine was obtained from Aladdin Macklin Biochemical Co., Ltd, Shanghai, China, nitric acid (HNO3), sulfuric acid (H2SO4), ammonia (NH3⋅H2O), and ethanol were obtained from Shanghai Chemical Reagent Co., Ltd., Shanghai, China. All the chemicals were used as obtained without any further purification. Deionized water was used throughout the experiments.
Preparations
Synthesis of g-C3N4 nanoparticles
g-C3N4 nanoparticles were synthesized by the acid etching method. Typically, 5 g melamine was put into a semi-enclosed crucible of aluminum oxide and heated at 600 °C for 2 h at the rate of 3 °C/min in air, therefore obtaining the bulk g-C3N4 (denoted as CN). Then, the bulk g-C3N4 powders were put into the mixture of nitric acid and sulfuric acid (the volume ratio of HNO3 to H2SO4 is 1:1) and refluxed at 80 °C for 2 h. Finally, the obtained powders were completely washed with distilled water and ethanol, and then dried at 70 °C for 8 h (denoted as CNNP). Then, the CNNP was ultrasonicated for 2 h and stirred for 12 h in deionized water to obtain the dispersion solution with the concentration of 10 mg/mL.
Synthesis of porous g-C3N4
Porous g-C3N4 (denoted as PCN) was directly prepared by the thermal polymerization strategy and the hydrothermal method. Firstly, in this experiment, the bulk g-C3N4 was prepared by the same procedure above. Then, 0.2 g of bulk g-C3N4 was dispersed in 50 mL distilled water and adjusted the pH value of the solution to 12 by using the ammonia. After continuously stirring for 30 min, the suspension was transferred into a 100 mL Teflon-lined autoclave. Subsequently, the autoclave was sealed, and heated at 140 °C for 12 h, then cooled down slowly. The resulting samples were filtered, washed thoroughly, and then dried at 60 °C overnight.
Fabrication of g-C3N4 nanoparticles/porous g-C3N4
The PCN was dispersed in distilled water and vigorously stirred for 30 min. After that, a certain amount of CNNP was added and ultrasonicated for 0.5 h, then was stirred for 12 h. Meanwhile, the precipitate was gathered and fully washed with deionized water, and then dried in an oven at 60 °C for 8 h. After heat treatment at 180 °C for 1 h, the g-C3N4 nanoparticles/porous g-C3N4 composite (denoted as CNNP@PCN) was obtained. For comparison, g-C3N4 nanoparticles/bulk g-C3N4 composite (denoted as CNNP@CN) was synthesized through the similar process.
Catalysts characterization
The crystal phase characteristics were measured by powder XRD (APEXII, Bruker, Karlsruhe, Germany) using Ni-filtered Cu Kα radiation with a step width of 0.02° and FTIR spectra which was revealed on a ThermoFisher Nicolet 5700 spectrometer (Massachusetts, USA) using the standard KBr disk method. XPS spectroscopy was obtained on a Thermo Fisher Escalab 250Xi (Massachusetts, USA) with the XPS survey spectra and high-resolution spectra. The morphologies and microstructures were directly revealed by SEM (SU-70, Hitachi, Tokyo, Japan) and TEM (Tecnai G2 F20 S-TWIN, FEI, Hillsboro, USA). The UV–Vis DRS (Cary 5000, Agilent, California, USA) were carried out using a BaSO4 disk as a reference. The Zeta potential was performed using a Zeta potential analyzer (Zetasizer Nano-ZS, Malvern, Malvern, UK). The BET surface area information of the as-prepared samples was recorded on a micromeritics ASAP 2020 N2 adsorption–desorption instrument. PL spectra and time-resolved photoluminescence (TR-PL) decay spectra were performed on a fluorescence spectrophotometer (FLS920, Edinburgh, UK) under the excitation wavelength of 365 nm. EPR (Bruker A300-10/12, Germany) signals were measured by the probe molecular 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) under visible-light irradiation. A 300 W Xe lamp with a 420 nm UV-cutoff filter provided the light source.
Evaluation of photocatalytic activity and adsorption capacity
The evaluation of the photocatalytic performance of CN, PCN, and CNNP@PCN composites was evaluated in aqueous solution by the degradation of MB. In each experiment, 50 mg of a photocatalyst was added into 100 mL of MB (10 mg/L) aqueous solution. Then, the mixture was magnetically agitated for 30 min in the dark to reach the equilibrium of adsorption and desorption. In the process of the photocatalytic reaction, 4 mL of the mixture was gathered every 30 min, and then centrifuged at a speed of 10,000 rpm for 8 min to eliminate the photocatalyst. Finally, a UV–Vis spectrophotometer (UV-4100, Hitachi, Japan) was taken measured to evaluate the concentration of MB, the maximum absorption wavelengths of which is at 664 nm. Adsorption experiments were taken measured in MB solution (100 mL) with an initial concentration of 10 mg/L. Then, 50 mg synthesized samples were added into the MB solution with continuous stirring in the dark. The as-prepared samples were taken at a certain interval time and centrifuged. The remaining test steps are similar to the above.
References
M.R. Hoffmann, S.T. Martin, W. Choi, and D.W. Bahnemann: Environmental applications of semiconductor photocatalysis. Chem. Rev. 95, 69 (1995).
A. Fujishima and K. Honda: Electrochemical photolysis of water at a semiconductor electrode. Nature 238, 37 (1972).
A.J. Bard: Photoelectrochemistry. Science 207, 139 (1980).
H. Xu, Z. Wu, and Y. Wang: Enhanced visible-light photocatalytic activity from graphene-like boron nitride anchored on graphitic carbon nitride sheets. J. Mater. Sci. 52, 9477 (2017).
C. Xu, J. Wang, and B. Gao: Synergistic adsorption and visible-light catalytic degradation of RhB from recyclable 3D mesoporous graphitic carbon nitride/reduced graphene oxide aerogels. J. Mater. Sci. 54, 8892 (2019).
J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. Lee, and J. Zhong: Metal-free efficient photocatalyst for stable visible water splitting via a two-electron pathway. Science 347, 970 (2015).
J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, and X. Fu: Sulfur-mediated synthesis of carbon nitride: Band-gap engineering and improved functions for photocatalysis. Energy Environ. Sci. 4, 675 (2011).
Z. Lin and X. Wang: Nanostructure engineering and doping of conjugated carbon nitride semiconductors for hydrogen photosynthesis. Angew. Chem. Int. Ed. 52, 1735 (2013).
X.C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, and J.M. Carlsson: A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nature Mater. 8, 76 (2009).
Y. Lu, B. Ma, and Y. Yang: High activity of hot electrons from bulk 3D graphene materials for efficient photocatalytic hydrogen production. Nano Res. 10, 1662 (2017).
Y. Luo, J. Wang, S. Yu, Y. Cao, K. Ma, Y. Pu, W. Zou, C. Tang, F. Gao, and L. Dong: Nonmetal element doped g-C3N4 with enhanced H2 evolution under visible light irradiation. J. Mater. Res. 33, 1268 (2018).
L. Peng, Z. Li, R. Zheng, H. Yu, and X. Dong: Preparation and characterization of mesoporous g-C3N4/SiO2 material with enhanced photocatalytic activity. J. Mater. Res. 34, 1785 (2019).
J. Wen, J. Xie, X. Chen, and X. Li: A review on g-C3N4-based photocatalysts. Appl. Surf. Sci. 391, 72 (2017).
Q. Li, X. Zhao, and J. Yang: Exploring the effects of nanocrystal facet orientations in g-C3N4/BiOCl heterostructures on photocatalytic performance. Nanoscale 7, 18971 (2015).
L. Sun, Y. Qi, and C.J. Jia: Enhanced visible-light photocatalytic activity of g-C3N4/Zn2GeO4 heterojunctions with effective interfaces based on band match. Nanoscale 6, 2649 (2014).
J. Shen, Y. Hui, and Q. Shen: Template-free preparation and properties of mesoporous g-C3N4/TiO2 nanocomposite photocatalyst. CrystEngComm 16, 1868 (2014).
D. Jiang, L. Chen, and J. Zhu: Novel p-n heterojunction photocatalyst constructed by porous graphite-like C3N4 and nanostructured BiOI: Facile synthesis and enhanced photocatalytic activity. Dalton Trans. 42, 15726 (2013).
K. Dai, L. Lu, and C. Liang: A high efficient graphitic-C3N4/BiOI/graphene oxide ternary nanocomposite heterostructured photocatalyst with graphene oxide as electron transport buffer material. Dalton Trans. 44, 7903 (2015).
J.S. Jang, S.J. Hong, and J.Y. Kim: Heterojunction photocatalyst TiO2/AgGaS2 for hydrogen production from water under visible light. Chem. Phys. Lett. 475, 78 (2009).
J.K. Yong, B. Gao, and Y.H. Song: Heterojunction of FeTiO3 nanodisc and TiO2 nanoparticle for a novel visible light photocatalyst. J. Phys. Chem. C 113, 19179 (2009).
J. Zou, Y. Yu, and W. Yan: A facile route to synthesize boron-doped g-C3N4 nanosheets with enhanced visible-light photocatalytic activity. J. Mater. Sci. 54, 6867 (2019).
J. Li, B. Shen, and Z. Hong: A facile approach to synthesize novel oxygen-doped g-C3N4 with superior visible-light photoreactivity. Chem. Commun. 48, 12017 (2012).
Z. Wang, W. Guan, Y. Sun, and F. Dong: Water-assisted production of honeycomb-like g-C3N4 with ultralong carrier lifetime and outstanding photocatalytic activity. Nanoscale 7, 7 (2015).
T. Li, L. Zhao, and Y. He: Synthesis of g-C3N4 /SmVO4 composite photocatalyst with improved visible light photocatalytic activities in RhB degradation. Appl. Catal. B Environ. 129, 255 (2013).
F.T. Li, Y. Zhao, and Q. Wang: Enhanced visible-light photocatalytic activity of active Al2O3/g-C3N4 heterojunctions synthesized via surface hydroxyl modification. J. Hazard. Mater. 283, 371 (2015).
X.J. Wang, L. Chao, and X.L. Li: Construction of g-C3N4/Al2O3 hybrids via in-situ acidification and exfoliation with enhanced photocatalytic activity. Appl. Surf. Sci. 394, 340 (2017).
Z. You, Y. Su, Y. Yu, H. Wang, T. Qin, F. Zhang, Q. Shen, and H. Yang: Preparation of g-C3N4 nanorod/InVO4 hollow sphere composite with enhanced visible-light photocatalytic activities. Appl. Catal. B Environ. 213, 127 (2017).
Y. Zhang and Z. Jin: Synergistic enhancement of hydrogen production by ZIF-67 (Co) derived Mo–Co–S modified g-C3N4/rGO photocatalyst. Catal. Lett. 149, 34 (2019).
D. Ren, W. Zhang, Y. Ding, R. Shen, Z. Jiang, X. Lu, and X. Li: In situ fabrication of robust cocatalyst-free CdS/g-C3N4 2D–2D step-scheme heterojunctions for highly active H2 evolution. Solar RRL. 1900423 (2019). doi:10.1002/solr.201900423.
Y. Zang, L. Li, X. Li, and R. Lin: Synergistic collaboration of g-C3N4/SnO2 composites for enhanced visible-light photocatalytic activity. Chem. Eng. J. 246, 277 (2014).
Z. Zhang, J. Huang, M. Zhang, and Q. Yuan: Ultrathin hexagonal SnS2 nanosheets coupled with g-C3N4 nanosheets as 2D/2D heterojunction photocatalysts toward high photocatalytic activity. Appl. Catal. B Environ. 163, 298 (2015).
L. Sun, X. Zhao, C.J. Jia, Y. Zhou, X. Cheng, P. Li, and L. Liu: Enhanced visible-light photocatalytic activity of g-C3N4-ZnWO4 by fabricating a heterojunction: Investigation based on experimental and theoretical studies. J. Mater. Chem. 22, 23428 (2012).
C. Pan, J. Xu, Y. Wang, and D. Li: Dramatic activity of C3N4/BiPO4 photocatalyst with core/shell structure formed by self-assembly. Adv. Funct. Mater. 22, 1518 (2012).
S. Kumar, T. Surendar, and A. Baruah: Synthesis of a novel and stable g-C3N4–Ag3PO4 hybrid nanocomposite photocatalyst and study of the photocatalytic activity under visible light irradiation. J. Mater. Chem. A 1, 5333 (2013).
B. Chai, F. Zou, and W. Chen: Facile synthesis of Ag3PO4/C3N4 composites with improved visible light photocatalytic activity. J. Mater. Res. 30, 1128 (2015).
F. Zhang, L. Wang, M. Xiao, F. Liu, X. Xu, and E. Du: Construction of direct solid-state Z-scheme g-C3N4/BiOI with improved photocatalytic activity for microcystin-LR degradation. J. Mater. Res. 33, 201 (2017).
S. Liu, J. Chen, D. Xu, X. Zhang, and M. Shen: Enhanced photocatalytic activity of direct Z-scheme Bi2O3/g-C3N4 composites via facile one-step fabrication. J. Mater. Res. 33, 1391 (2018).
X. Yan, X. Yuan, J. Wang, Q. Wang, C. Zhou, D. Wang, H. Tang, J. Pan, and X. Cheng: Construction of novel ternary dual Z-scheme Ag3VO4/C3N4/reduced TiO2 composite with excellent visible-light photodegradation activity. J. Mater. Res. 34, 2024 (2019).
X. Lu, J. Xie, X. Chen, and X. Li: Engineering MPx (M = Fe, Co or Ni) interface electron transfer channels for boosting photocatalytic H2 evolution over g-C3N4/MoS2 layered heterojunctions. Appl. Catal. B Environ. 252, 250 (2019).
X. Lu, J. Xie, S.-y. Liu, A. Adamski, X. Chen, and X. Li: Low-cost Ni3B/Ni(OH)2 as an ecofriendly hybrid cocatalyst for remarkably boosting photocatalytic H2 production over g-C3N4 nanosheets. ACS Sustain. Chem. Eng. 6, 13140 (2018).
R. Shen, W. Liu, D. Ren, J. Xie, and X. Li: Co1.4Ni0.6P cocatalysts modified metallic carbon black/g-C3N4 nanosheet Schottky heterojunctions for active and durable photocatalytic H2 production. Appl. Surf. Sci. 466, 393 (2019).
D. Jiang, J. Li, C. Xing, Z. Zhang, and S. Meng: Two dimensional CaIn2S4/g-C3N4 heterojunction nanocomposite with enhanced visible-light photocatalytic activities: Interfacial engineering and mechanism insight. ACS Appl. Mater. Interfaces 7, 19234 (2015).
J. Wang, L. Tang, G. Zeng, Y. Deng, Y. Liu, L. Wang, Y. Zhou, Z. Guo, J. Wang, and C. Zhang: Atomic scale g-C3N4/Bi2WO6 2D/2D heterojunction with enhanced photocatalytic degradation of ibuprofen under visible light irradiation. Appl. Catal. B Environ. 209, 285 (2017).
W. Yu, J. Chen, T. Shang, L. Chen, L. Gu, and T. Peng: Direct Z-scheme g-C3N4/WO3 photocatalyst with atomically defined junction for H2 production. Appl. Catal. B Environ. 219, 693 (2017).
J. Xiao, W. Huang, Y. Hu, F. Zeng, Q. Huang, B. Zhou, A. Pan, K. Li, and G. Huang: Facile in situ synthesis of wurtzite ZnS/ZnO core/shell heterostructure with highly efficient visible-light photocatalytic activity and photostability. J. Phys. D Appl. Phys. 51, 075501 (2018).
S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, and X. Wang: Exfoliated graphitic carbon nitride nanosheets as efficient catalysts for hydrogen evolution under visible light. Adv. Mater. 25, 2452 (2013).
Y. Feng, J. Shen, Q. Cai, and H. Yang: The preparation and properties of a g-C3N4/AgBr nanocomposite photocatalyst based on protonation pretreatment. New J. Chem. 39, 1132 (2015).
Z. Feng, L. Zeng, Y. Chen, Y. Ma, C. Zhao, R. Jin, Y. Lu, Y. Wu, and Y. He: In situ preparation of Z-scheme MoO3/g-C3N4 composite with high performance in photocatalytic CO2 reduction and RhB degradation. J. Mater. Res. 32, 3660 (2017).
X. Zhang, H. Wang, H. Wang, Q. Zhang, J. Xie, Y. Tian, and J. Wang: Single-layered graphitic-C3N4 quantum dots for two-photon fluorescence imaging of cellular nucleus. Adv. Mater. 26, 4438 (2014).
S. Wang, C. Li, T. Wang, P. Zhang, and A. Li: Controllable synthesis of nanotube-type graphitic C3N4 and their visible-light photocatalytic and fluorescent properties. J. Mater. Chem. A 2, 2885 (2014).
M. Hua, Z. Xing, Y. Cao, Z. Li, X. Yan, Z. Xiu, T. Zhao, S. Yang, and W. Zhou: Ti3+ self-doped mesoporous black TiO2/SiO2/g-C3N4 sheets heterojunctions as remarkable visible-light driven photocatalysts. Appl. Catal. B Environ. 226, 499 (2018).
J. Zhang, J. Fu, Z. Wang, B. Cheng, K. Dai, and W. Ho: Direct Z-scheme porous g-C3N4/BiOI heterojunction for enhanced visible-light photocatalytic activity. J. Alloys Compd. 766, 841 (2018).
S. Zhang, J. Li, X. Wang, Y. Huang, and M. Zeng: In situ ion exchange synthesis of strongly coupled Ag@AgCl/g-C3N4 porous nanosheets as plasmonic photocatalyst for highly efficient visible-light photocatalysis. ACS Appl. Mater. Interfaces 6, 22116 (2014).
P. Niu, L. Zhang, G. Liu, and H.M. Cheng: Graphene-like carbon nitride nanosheets for improved photocatalytic activities. Adv. Funct. Mater. 22, 4763 (2012).
D.R. Paul, R. Sharma, S.P. Nehra, and A. Sharma: Effect of calcination temperature, pH and catalyst loading on photodegradation efficiency of urea derived graphitic carbon nitride towards methylene blue dye solution. RSC Adv. 9, 15381 (2019).
L. Heymann, B. Schiller, H. Noei, A. Stierle, and C. Klinke: A new synthesis approach for carbon nitrides: Poly(triazine imide) and its photocatalytic properties. ACS Omega 3, 3892 (2018).
T. Wang, M. Huang, X. Liu, Z. Zhang, Y. Liu, W. Tang, S. Bao, and T. Fang: Facile one-step hydrothermal synthesis of α-Fe2O3/g-C3N4 composites for the synergistic adsorption and photodegradation of dyes. RSC Adv. 9, 29109 (2019).
Y. Li, Z. Li, and L. Gao: Construction of Z-scheme BiOI/g-C3N4 heterojunction with enhanced photocatalytic activity and stability under visible light. J. Mater. Sci. Mater. Electron. 30, 12769 (2019).
X. Li, J. Xie, C. Jiang, J. Yu, and P. Zhang: Review on design and evaluation of environmental photocatalysts. Front. Environ. Sci. Eng. 12, 14 (2018).
F. Wu, X. Li, W. Liu, and S. Zhang: Highly enhanced photocatalytic degradation of methylene blue over the indirect all-solid-state Z-scheme g-C3N4-RGO-TiO2 nanoheterojunctions. Appl. Surf. Sci. 405, 60 (2017).
Acknowledgments
This work is supported by the National Key Research and Development Program of China (No. 2018YFD1101003).
Author information
Authors and Affiliations
Corresponding author
Supplementary Material
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1557/jmr.2020.182.
Rights and permissions
About this article

Cite this article
Shen, Q., Wu, C., You, Z. et al. g-C3N4 nanoparticle@porous g-C3N4 composite photocatalytic materials with significantly enhanced photo-generated carrier separation efficiency. Journal of Materials Research 35, 2148–2157 (2020). https://doi.org/10.1557/jmr.2020.182
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/jmr.2020.182