
Abstract
The particle size of Fe3O4 nanoparticles is controlled using a simple oxidation–precipitation method without any surfactant. The structure, morphology and physical properties of the synthesized Fe3O4 NPs were characterized using x-ray diffraction, scanning electron microscopy, transmission electron microscopy, x-ray photoelectron spectroscopy, Brunauer–Emmett–Teller, and vibrating sample magnetometer. As-prepared magnetite samples exhibited spherical morphology with average diameters of 30, 70, 250, and 600 nm, respectively. Activity of the synthesized Fe3O4 NPs was evaluated for the Fenton-like reaction, using rhodamine B (RhB) as a model molecule. The results showed that catalytic activity increases with the reduced particle size. The significant higher catalytic activity of the fine Fe3O4 NPs mainly originated from the higher specific surface area, due to the increase in exposed active site number and adsorption capacity. The reusability of 30 nm Fe3O4 NPs was also investigated after three successive runs, in which the RhB degradation performances showed a slight difference with the first oxidation cycle. This investigation is of great significance for the promising application of the heterogeneous Fenton catalyst with enhanced activity in the oxidative degradation of organic pollutants.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
I. INTRODUCTION
As one of the important contaminants of water pollution, the refractory organics have harmful effects on human beings and environment. Since refractory organic pollutants are highly hazardous and suspicious carcinogens, the stringent water quality regulations are imposed and wastewater containing these chemicals must be treated accordingly. Recently, various technologies including biological degradation, physical adsorption and chemical oxidation have been developed to remove the aqueous pollutants. Among these methods, advanced oxidation processes, heterogeneous Fenton process with the capability of exploiting the high reactivity of hydroxyl radicals (˙OH) in driving oxidation, emerged a promising technology for the degradation of refractory organic pollutants.1,2 Various heterogeneous catalysts including iron minerals,3 zero-valent iron,4 soils,5 and carbon materials6 have been prepared and tested for the Fenton-like oxidation of model pollutants and practical wastewaters to find the new catalytic materials with high activity and stability. Among these materials, magnetite (Fe3O4) has received much more attention due to its low cost, easy separation, environment-friendly property, strong adsorption capacity, and enhanced stability.7,8 Since Fe3O4 nanoparticles (NPs) were found to possess intrinsic peroxidase-like activity,9 magnetite has been widely explored for the applications in the degradation of various refractory organics such as phenol,10 pentachlorophenol,11p-nitrophenol,12 2,4-dichlorophenol13 and dye pollutants.14 The catalytic mechanism for the activation of H2O2 by Fe3O4 NPs as the heterogeneous Fenton catalyst is shown in Eqs. (1)–(6).11,14
The unique crystal structure of Fe3O4 allows the Fe species to be reversibly oxidized and reduced while keeping the same structure, which makes the magnetite to be an excellent catalyst for Fenton-like processes. However, several reports demonstrated that the H2O2-activating ability of Fe3O4 NPs was not so strong for the removal of refractory organic pollutants from practical wastewaters.14,15 Furthermore, with a proper control of crystallinity and structure of the Fe3O4 NPs, the functional performance can be strongly improved.
It is well accepted that the particle size of Fe3O4 NPs has a strong effect on their intrinsic properties.16–18 The particle size of Fe3O4 NPs is generally controlled by the synthesis method. As far, a wide variety of methods to synthesize Fe3O4 NPs have been reported, including hydrothermal method,19 thermal decomposition,20 sol–gel method,21 and chemical precipitation.22 In practical industrial application, as well as large scale production, the chemical precipitation is considered to be the simplest, most efficient, and economic pathway to obtain magnetic particles. However, there has been few attempts to achieve size-controlled synthesis of Fe3O4 NPs through a simple and economic pathway. Moreover, little is known about the effects of particle sizes in Fenton-like catalytic activity of Fe3O4 NPs.
Herein, we described the size-controllable synthesis of Fe3O4 NPs under a simple and efficient oxidation–precipitation procedure without any surfactant. Using this method, the concentration, pH and the ratio of Fe2+/Fe3+ could be exactly controlled, which might be responsible for the formation of different sizes of Fe3O4 NPs.23–25 As we known, rhodamine B (RhB) is one of the most commonly used xanthenes dyes for the textile industry. The RhB can well dissolve in water or organic solvent with many toxic effects such as irritation to the skin, eyes, and respiratory tracts. Even more serious, RhB was believed to be carcinogenic, which made it be banned from using in foods and cosmetics. However, the RhB is still extensively used in dyeing and staining processes.26 Therefore, an efficient treatment of effluents containing RhB is essential before their final discharge into the aquatic environment. Besides, RhB has been selected as a model dyeing pollutant in numerous researches on Fenton reaction.14,27–30 In this study, the synthesized Fe3O4 NPs of various sizes were used as heterogeneous Fenton catalysts for adsorbing and degrading RhB in the presence of H2O2. The physical and chemical characterization of Fe3O4 was performed and the structural effects on their Fenton-like catalytic activity were investigated, which is of great importance for obtaining a profound understanding of catalytic processes, as well as for improving the catalytic properties of materials.
II. EXPERIMENTAL SECTION
A. Synthesis of the Fe3O4 NPs
The Fe3O4 NPs of various sizes were synthesized by oxidation–precipitation method. Ferrous sulfate (FeSO4) was used as iron salts, and sodium nitrate (NaNO3) was used as the oxidizing agent. All chemical reagents were of analytical grade and used as received. In all experiments, distilled water was used for preparing the solutions. In a typical procedure for the fabrication of Fe3O4 NPs with diameters of approximately 600 nm (denoted as sample d), 3.2 g of NaOH was dissolved in 100 mL distilled water after using N2 to lustrate oxygen in it for 20 min. 100 mL of 0.5 M FeSO4 aqueous solution was added dropwise into the flask under stirring and N2 flow. The solution was subsequently placed into a 95 °C water bath. Afterward, 50 mL of 0.5 M NaNO3 aqueous solution was added dropwise (10 mL/min) into the suspension and the reaction was kept for 4 h. Vigorous stirring and stable N2 flow were kept during the entire experiment. After cooled to room temperature, the formed Fe3O4 NPs were collected under the assistance of a magnet and washed five times with distilled water and absolute ethanol under ultrasonication, and then dried in a vacuum oven at 100 °C for 12 h.
When NaNO3 was increased 3-fold and reaction time was reduced by half, we added the NaOH aqueous solution dropwise into the mixed solution of FeSO4 and NaNO3 in the above procedures while keeping other parameters unchanged, 250 nm Fe3O4 (denoted as sample c) NPs were obtained. Similarly, when FeSO4 aqueous solution was added dropwise into the mixed solution of NaOH and NaNO3 in the procedures of fabrication of 250 nm Fe3O4 NPs without changing other parameters, 70 nm Fe3O4 NPs (denoted as sample b) were synthesized. 30 nm Fe3O4 NPs (denoted as sample a) were obtained similarly with the 70 nm Fe3O4 NPs when the concentration of reactants was reduced 3-fold. All the products were stored in desiccator under ambient temperature for further experiments.
B. Characterization
The structure and morphology of the Fe3O4 samples were characterized by x-ray powder diffractometer (XRD; Rigaku D/max-RB, Tokyo, Japan) with Cu Kα radiation, scanning electron microscopy (SEM; FEI Nova400, FEI Company, Brno, Czech Republic) and transmission electron microscopy (TEM; JEOL JEM-100CXII, Tokyo, Japan). High resolution TEM images were obtained by using JEOL JEM-2100F microscope operated at an acceleration voltage of 100 kV. Magnetic characterization of the samples was performed using a JDAW-2000D vibrating sample magnetometer (Yingpu Corp., Hangzhou, China) at room temperature. N2 adsorption–desorption isotherms of the samples were recorded on an ASAP 2020-M micropore physisorption analyzer (Micromeritics, Atlanta, Georgia). Specific surface areas were calculated by the Brunauer–Emmett–Teller (BET) method. The bulk ratio between FeII and FeIII in the Fe3O4 was determined by 1,10-phenanthroline spectrophotometric method after the samples were dissolved in acidic media at 60 °C for 48 h under a nitrogen atmosphere.5,11 The surface ratio of FeII/FeIII on the Fe3O4 surface was analyzed by x-ray photoelectron spectroscopy (XPS; VG Multilab 2000, Thermo Fisher Scientific, Waltham, Massachusetts). Curve fittings were achieved with the XPSPeak 4.1 program using the Fe 2p binding energies region for the analysis.
C. Sorption experiments
To determine the adsorption performance of the obtained Fe3O4 NPs toward organic pollutant, sorption isotherm experiment was carried out at room temperature (25 °C) in the dark. The solid samples were mixed with aqueous solutions of RhB at different initial concentrations, stirring 12 h to ensure the adsorption equilibrium. Before analysis, the suspensions were magnetically separated and the clear water phase was decanted. Then the equilibrium concentrations of RhB in the aqueous phase were measured and sorbed concentrations were calculated according to:
where, qe (mg/g), Ci (mg/L), Ce (mg/L), Ms (g), and V (L) are the sorbed RhB concentration, initial concentration, equilibrium concentration, sorbent dosage, and volume of the solution, respectively.
D. Fenton-like catalytic activity test
The Fenton-like catalytic activity test was performed under room temperature (25 °C) in a 250 mL glass reactor (containing 100 mL of reaction solution) continuously stirred using RhB (Shanghai Zhanyun Chemical Co., Ltd., Shanghai, China) as reactant and H2O2 as oxidant. The reaction pH value was adjusted to 3.0, the optimum pH for the typical Fenton process, before the oxidation.3,10 Before reaction, the suspension containing Fe3O4 (1 g/L) and RhB (40 mg/L) was stirred for 2 h to achieve adsorption equilibrium. The degradation reaction was initiated when H2O2 (50 mM) was added to the solution. During all the oxidation reactions, 5 mL aliquots were withdrawn and clarified quickly by a strong outer permanent magnet at selected time intervals. The aqueous phase was sampled for analysis. The solid catalyst separated from the aqueous phase was rinsed by 5 mL distilled water for three times. The rinsed liquid was mixed for analysis. The residual RhB amount is the sum of that in the aqueous and solid phase. The concentration of RhB was analyzed by UV–vis spectroscopy (Ultrospec 3300 pro, GE Healthcare Bio-Sciences China Ltd., Shanghai, China) at a wave length of 554 nm.28 The chemical oxygen demand (COD) was measured by the dichromate method. At the end of the catalytic test, the concentration of iron leaching in the solution was measured by the 1,10-phenanthroline spectrophotometric method. All experimental runs were performed in the dark.
III. RESULTS AND DISCUSSION
A. Characterization of the synthesized Fe3O4 NPs
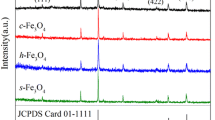
X-ray diffraction (XRD) was carried out to identify the structures and crystallinity of the synthesized Fe3O4 NPs. As shown in Fig. 1, all of the obtained products show peaks at 18.3, 30.1, 35.5, 43.1, 53.5, 57.0, and 62.6°, which can be indexed to the reflections from crystal planes (111), (220), (311), (400), (422), (511), and (440) of the cubic phase of Fe3O4 according to the standard JCPDS data (card no. 01–1111).31 The absence of any unidentified peaks in Fig. 1 indicates that the synthesized NPs are pure and crystalline. Additionally, there was a difference in XRD peak intensity among the XRD patterns of the four Fe3O4 NPs samples. The application of Scherrer equation to the (311) reflection peak indicated that the crystal sizes were 21.7, 39.5, 60.8, and 80.5 nm for the four Fe3O4 NPs samples, as listed in Table I.

XRD patterns of the synthesized Fe3O4 NPs: (a) 30 nm, (b) 70 nm, (c) 250 nm, and (d) 600 nm.
The size and morphology of the four Fe3O4 NPs samples were further studied with SEM and TEM. As can be seen from Fig. 2, the four Fe3O4 NPs samples exhibit spherical morphologies with average particle sizes of approximately 30, 70, 250, and 600 nm, respectively, which are much larger than the crystal size obtained from XRD analysis. The main reason is that the Fe3O4 NPs were the results of secondary growth of crystal grain. The crystal sizes obtained from XRD results represent the sizes of Fe3O4 crystal grains, while the average particle sizes observed from SEM and TEM show the sizes of Fe3O4 NPs. This may be due to the synthesis method of Fe3O4 NPs.32 As reported, the formation of Fe3O4 follows the nucleation-growth mechanism through oxidation–precipitation method. The nucleation of magnetite particles occurs when the molar ratio of Fe(OH)2/Fe(OH)3 becomes close to 1:2, which is the characteristic value for the magnetite structure. Then the core of Fe3O4 begins to grow up.32 Therefore, the sizes of the product were strongly depend on reaction conditions such as reaction time, molar ratio of FeSO4 and oxidizing agent, and the concentration of FeSO4, etc. The possible mechanism of formation of Fe3O4 can be described as follows33:

SEM and TEM images of the synthesized Fe3O4 NPs: (a-1, a-2) 30 nm, (b-1, b-2) 70 nm, (c-1, c-2) 250 nm, and (d-1, d-2) 600 nm, and HR-TEM images of 30 nm Fe3O4 NPs (e-1, e-2).
In the procedures of fabrication of 600 nm Fe3O4 NPs [Fig. 2(d)], the FeSO4 was first added into the NaOH solution to form a milk white Fe(OH)2 suspension. After the addition of NaNO3, the suspension gradually turned dark green then black in color, which indicated the formation of Fe3O4. However, when we increased 3 times of the NaNO3 dosage and changed the order of adding reagents (adding NaOH into the mixture of FeSO4 and NaNO3), the ratio of ferrous to ferric ions in the metal salt solution was decreased. Thus, the smaller hydroxide particles would be generated readily, resulting in the formation of small Fe3O4 NPs.23 On the other hand, the reaction time was shortened from 4 to 2 h, which further made the Fe3O4 NPs grow smaller.24 Thus, the Fe3O4 NPs with smaller dimension [250 nm, Fig. 2(c)] were synthesized using a lower molar ratio of FeSO4 and oxidizing agent and a shorter growth time. When we further changed the order of adding reagents (adding FeSO4 into the mixture of NaOH and NaNO3), a black precipitate of Fe3O4 NPs was formed in a very short time after mixing the iron salt solution with the base solution, which indicated that little intermediates were formed prior to the formation of Fe3O4. This observation demonstrates that the reaction mechanism for the formation of Fe3O4 NPs may be different between the two cases, which may influence the size of Fe3O4 NPs. The formation of Fe3O4 NPs in this case is more likely by the “coprecipitation” reaction,23 which is supposed to be responsible for the formation of smaller Fe3O4 NPs [70 nm, Fig. 2(b)]. Several papers have reported that the size of the synthesized Fe3O4 NPs increases with the increase of the initial ferric salt concentration.25,34 In the present study, we have similar results. When the ferric salt concentration was reduced 3-fold, Fe2+ was kept at a low concentration and could be easily oxidized to Fe3+. As a result, more crystal nuclei were generated in the solution, which led to a small particle size [30 nm, Fig. 2(a)]. Moreover, further characterization of crystallinity by HR-TEM was conducted on the 30 nm Fe3O4 NPs. The HR-TEM images shown in Fig. 2(e) demonstrate that highly crystalline Fe3O4 NPs with lattice fringes were formed in the synthesis process, which correspond to (111) plane of Fe3O4 crystal with lattice spacing of 0.485 nm. In general, we are able to prepare satisfying Fe3O4 NPs with controllable sizes by varying the experimental conditions through a simple oxidation–precipitation method with any surfactant.
Magnetic properties of the obtained Fe3O4 NPs were characterized by measuring the applied field dependence of magnetization (Fig. 3). The values of saturation magnetization (Ms) and coercivity (Hc) read from the magnetization curves are tabulated in Table I. As can be seen, the Ms and Hc of the obtained Fe3O4 NPs decreased with the decrease of particle sizes, which may be due to the formation of the magnetically disordered surface as a result of the large surface-to-volume ratio.35 However, the value of Ms of the 30 nm Fe3O4 NPs was still as high as 80.9 emu/g, which are found to be 87% of the value of bulk Fe3O4 (93 emu/g).23 Moreover, room-temperature magnetic response of the 30 nm Fe3O4 NPs was also examined by an external magnet (Fig. 3 inset). Upon placement of an external magnet beside the sample, the Fe3O4 NPs quickly concentrated on the side, leaving the solution transparent, which demonstrated the excellent magnetic response of the synthesized Fe3O4 NPs to a magnetic field.

Hysteresis loop of the synthesized Fe3O4 NPs.
Nitrogen adsorption/desorption isotherms of the obtained Fe3O4 NPs showed in Fig. 4 display type II isotherms. The hysteresis area of N2 adsorption/desorption isotherms suggest the occurrence of capillary condensation and the wide distributions of pores in Fe3O4 NPs. The specific surface areas obtained from N2 adsorption measurements for the corresponding samples were 42.64, 39.51, 6.39, and 2.86 m2/g, respectively, as presented in Table I. Notably, the sample exhibited a smaller crystal size showed larger specific surface area, which was also found in other reports.14,18 In addition, FeII/FeIII ratios of the obtained Fe3O4 NPs were also measured in the bulk by chemical analysis, which are presented in Table I. As can be seen, the FeII/FeIII ratios obtained over the synthesized Fe3O4 NPs were close to the theoretical FeII/FeIII ratio (0.5) for Fe3O4 materials.

Nitrogen adsorption/desorption isotherms for the synthesized Fe3O4 NPs.
B. Adsorption isotherm
According to previous studies, the adsorption performance of the catalysts has a strong effect on their catalytic activity.36 Thus, the adsorption isotherms were carried out to investigate the contaminant sorption on the obtained Fe3O4 NPs (Fig. 5). RhB aqueous solutions with different initial concentrations varying from 5 mg/L to 400 mg/L were used for the adsorption experiment. The experimental isotherm data were fitted to the equations of Langmuir and Freundlich by applying linear regression analysis. On the basis of the statistic analysis, the isotherms can be well described with the Langmuir model which assumed that the single adsorbate binds to a single site on the adsorbent and that all surface sites on the adsorbents have the same affinity for the adsorbate and no interactions between the adsorbates. The linear form of the Langmuir equation is given by:
where qm (mg g−1) is the monolayer adsorption capacity, and Ka (L mg−1) is the Langmuir constant. The fitted Langmuir equations are qe = 34.64 × 0.029Ce/(1 + 0.029Ce) (R2 = 0.9962), qe = 27.79 × 0.033Ce/(1 + 0.033Ce) (R2 = 0.9849), qe = 5.44 × 0.027Ce/(1 + 0.027Ce) (R2 = 0.9908) and qe = 3.49 × 0.032Ce/(1 + 0.032Ce) (R2 = 0.9898) for the 30, 70, 250, and 600 nm Fe3O4 NPs, respectively. Good correlations imply that the adsorption of RhB on the catalysts is monolayer adsorption. The adsorption capacities of RhB onto the obtained Fe3O4 NPs are listed in Table I. It is obviously shown that the adsorption capacity of Fe3O4 NPs increased with decreasing particle size or increasing specific surface area. As the average particle size decreased to 30 nm, the specific surface area and adsorption capacity reached as high as 42.64 m2/g and 34.64 mg/g, respectively, which are almost fifteen and ten times higher than that for the 600 nm Fe3O4 samples.

Isothermal adsorption of RhB on the synthesized Fe3O4 NPs ([adsorbent] = 1 g/L; T = 25 °C).
C. The catalytic activity of the synthesized Fe3O4 NPs
The catalytical degradation efficiencies of RhB by different sizes of Fe3O4 NPs were shown in Fig. 6(a). The ability of H2O2 for the elimination of RhB without any heterogeneous catalyst was also evaluated. As shown in Fig. 6(a), the RhB conversion is almost negligible in the presence of H2O2 only, which may be due to the low oxidation potential of H2O2 compared with ˙OH.2 When the catalyst was added to the RhB solution, a significant RhB reduction was achieved. The RhB conversion rates of 30, 70, 250, and 600 nm Fe3O4 NPs were 96.3, 94.3, 73.5, and 49.0% after 300 min, respectively. Many reports have indicated that ˙OH could be generated by the reaction between H2O2 and Fe3O4 leading to the degradation of RhB, which has been described in Eqs. (1)–(6). The degradation of RhB in the Fe3O4/H2O2 system follows the pseudo-first-order kinetics, as given below:
where C0 and Ct are the concentrations of RhB at the beginning and time t, respectively, and k is the apparent rate constant (min−1). The representative plots of −ln(Ct/C0) versus t are presented in Fig. 6(b) and good correlation coefficients (R2 ∼ 0.99) are obtained in our system. As evident in Fig. 6 and Table II, it is obvious that the 30 nm Fe3O4 NPs exhibited superior catalytic activity in the Fe3O4/H2O2 process compared with the other sizes of Fe3O4. The results in Fig. 6, Table I, and Table II then indicated that a greater specific surface area or adsorption capacity was beneficial to the heterogeneous Fenton-like reaction, leading to a high degradation degree of RhB. Figure 7 shows the evolution of the apparent rate constant with the specific surface area. It can be obviously seen that the catalytic activity of Fe3O4 NPs increased with increasing specific surface area. The results indicated that the Fenton-like process in Fe3O4/H2O2 system was a surface reaction since a larger specific surface area provided more active sites, which lead to more efficient contact with the substrate. Higher content of surface ˙OH could be produced over high specific surface area Fe3O4 NPs, enhancing the catalytic oxidation of RhB. On the other hand, the Fe3O4 NPs with smaller particle size could adsorb more RhB molecules on the surface (Table I), which were easily attacked by the generated ˙OH, resulting in higher removal efficiency. Similar situations have also been observed in several works that it is the adsorption property of heterogeneous catalyst caused the improvement in the rate of substrate degradation.7,8,36 It is well accepted that the Fenton-like oxidation by Fe3O4 NPs is controlled by surface mechanism reaction and adsorption of H2O2 or RhB on the surface of magnetite can affect the whole degradation reaction rate.11 Compared with the 200 or 600 nm Fe3O4 NPs, there are more active sites (such as FeII and FeIII) exposed on the surface of 30 nm Fe3O4 NPs, resulting in more active radicals generated according to Eqs. (1)–(4). Meanwhile, more RhB molecules were adsorbed on the surface of 30 nm Fe3O4 NPs. Thus, the reaction between active radicals and RhB on the surface of 30 nm Fe3O4 NPs was accelerated rapidly. Therefore, the significant higher catalytic activity of the fine Fe3O4 NPs mainly originated from the higher specific surface area, as demonstrated by Fig. 7, due to the increase in exposed active site number and adsorption capacity.

RhB removal efficiency (a) and pseudo-first-order rate (b) of the synthesized Fe3O4 NPs of different sizes (C0 = 40 mg/L, [catalyst] = 1 g/L, [H2O2] = 50 mM, T = 25 °C, pH = 3).

The relationship between the constant rate and specific surface area of the Fe3O4 NPs of different sizes.
Furthermore, we also examined the contribution of homogeneous Fenton reaction catalyzed by dissolved iron to the whole degradation process. Hence, a special experiment was conducted after the degradation experiment. The solid catalysts were removed from the reaction solution by a Millipore filter (pore size 0.22 μm), then RhB and H2O2 were added to the filtrate. The operation of homogeneous Fenton reaction catalyzed by dissolved iron was similar to that of heterogeneous reaction mentioned in Sec. II. The results showed in Fig. S1 in the supplementary data revealed that the RhB conversion through homogeneous Fenton reaction was below 10% after 300 min. Thus, it indicated that the effect of dissolved iron on the degradation process is limited, which further demonstrated that the whole degradation process of RhB was a surface Fenton-like reaction. Moreover, the dissolved iron concentration was also measured after the reaction (Table II). The Fe leaching did not exceed 0.3 mg/L of each sample, which were negligible.
D. Reusability and stability of the synthesized Fe3O4 NPs
To investigate the reusability of the synthesized Fe3O4 NPs as heterogeneous catalysts, the recycling catalysis experiments of the 30 nm Fe3O4 NPs were carried out and the catalytic activity was examined in each cycle. After each recycling, the catalyst was treated by magnetic separation, dried and reused. As shown in Fig. 8 and Table II, only a small decrease in the RhB conversion and COD removal were observed after three successive cycles, with no significant reduction as a whole. XPS was used to quantify the changes of surface FeII/FeIII ratio. The details of the Fe 2p peaks of the 30 nm Fe3O4 before and after use during the RhB degradation are presented in Fig. 9. The peaks located at about 710.9 and 724.1 eV represented the binding energies of Fe 2p3/2 and Fe 2p1/2, respectively, and the binding energies of 709.9 and 711.2 eV indicated the presence of FeII and FeIII, consistent with the literature data for magnetite.14,37 With the help of peak shape parameters, the Fe 2p1/2 peak for Fe3O4 was deconvoluted into FeII and FeIII component, which is found to be 37.2 and 62.8% of the total iron surface atoms, respectively. For the sample after RhB degradation, the FeII is found to be 27.5% and FeIII 72.5%. It is obvious that parts of the FeII in the outermost layer of Fe3O4 are oxidized into FeIII during Fenton oxidation reaction, which may lead to the slight decrease in the removal efficiency of RhB between the three successive cycles. However, the RhB conversion and COD removal still remained high (Table II), which indicated that satisfying catalytic activity was maintained after three runs even if the Fe3O4 was observed to slightly dissolve at each cycle (the Fe leaching was 0.17–0.26 mg/L at the end of each test, Table II). In addition, TEM images (Fig. S2) and XRD patterns (Fig. S3) of the Fe3O4 NPs after reaction in the supplementary data showed that the morphology, dimension, and structure of the Fe3O4 NPs remained unchanged, and no clear agglomeration was observed. These results indicate that the synthesized Fe3O4 NPs were stable and exhibited excellent reusability in the catalysis process.

Effect of recycling the catalyst on the removal of RhB. Inset: fitted by pseudo-first-order rate law (C0 = 40 mg/L, [30 nm Fe3O4] = 1 g/L, [H2O2] = 50 mM, T = 25 °C, pH = 3).

XPS spectrum of Fe on 30 nm Fe3O4 NPs surface before and after degradation of RhB (Fe 2p line).
IV. CONCLUSIONS
Fe3O4 NPs, which exhibit spherical morphology with different particle sizes (30, 70, 250, and 600 nm), have been successfully fabricated by a simple oxidation–precipitation method without any surfactant. The synthesized Fe3O4 NPs are well-crystallized, as well as uniform and well-distributed. The Fenton-like catalytic activities of these Fe3O4 NPs vary substantially in the order 30 nm > 70 nm > 250 nm > 600 nm. The significant higher catalytic activity of the fine Fe3O4 NPs mainly originated from the higher specific surface area, due to the increase in exposed active site number and adsorption capacity. Stability studies evidenced satisfying stability of the synthesized Fe3O4 NPs, with limited loss in catalytic activity. This investigation is of particular significance for profound understanding and practical application of heterogeneous Fenton catalysts for the oxidative degradation of organic compounds.
References
K.H. Kim and S.K. Ihm: Heterogeneous catalytic wet air oxidation of refractory organic pollutants in industrial wastewaters: A review. J. Hazard. Mater. 186 (1), 16 (2011).
P.V. Nidheesh: Heterogeneous Fenton catalysts for the abatement of organic pollutants from aqueous solution: A review. RSC Adv. 5 (51), 40552 (2015).
R. Matta, K. Hanna, and S. Chiron: Fenton-like oxidation of 2,4,6-trinitrotoluene using different iron minerals. Sci. Total Environ. 385 (1–3), 242 (2007).
L. Xu and J. Wang: A heterogeneous Fenton-like system with nanoparticulate zero-valent iron for removal of 4-chloro-3-methyl phenol. J. Hazard. Mater. 186 (1), 256 (2011).
L. Chen, C. Deng, F. Wu, and N. Deng: Decolorization of the azo dye Orange II in a montmorillonite/H2O2 system. Desalination 281, 306 (2011).
L. Gu, N. Zhu, H. Guo, S. Huang, Z. Lou, and H. Yuan: Adsorption and Fenton-like degradation of naphthalene dye intermediate on sewage sludge derived porous carbon. J. Hazard. Mater. 246–247, 145 (2013).
S. Song, H. Yang, R. Rao, H. Liu, and A. Zhang: High catalytic activity and selectivity for hydroxylation of benzene to phenol over multi-walled carbon nanotubes supported Fe3O4 catalyst. Appl. Catal., A 375 (2), 265 (2010).
G.M. Ucoski, F.S. Nunes, G. DeFreitas-Silva, Y.M. Idemori, and S. Nakagaki: Metalloporphyrins immobilized on silica-coated Fe3O4 nanoparticles: Magnetically recoverable catalysts for the oxidation of organic substrates. Appl. Catal., A 459, 121 (2013).
L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett, and X. Yan: Intrinsic peroxidase-like activity of ferromagnetic nanoparticles. Nat. Nanotechnol. 2 (9), 577 (2007).
J. Zhang, J. Zhuang, L. Gao, Y. Zhang, N. Gu, J. Feng, D. Yang, J. Zhu, and X. Yan: Decomposing phenol by the hidden talent of ferromagnetic nanoparticles. Chemosphere 73 (9), 1524 (2008).
X. Xue, K. Hanna, M. Abdelmoula, and N. Deng: Adsorption and oxidation of PCP on the surface of magnetite: Kinetic experiments and spectroscopic investigations. Appl. Catal., B 89 (3–4), 432 (2009).
S.P. Sun and A.T. Lemley: p-Nitrophenol degradation by a heterogeneous Fenton-like reaction on nano-magnetite: Process optimization, kinetics, and degradation pathways. J. Mol. Catal. A: Chem. 349 (1–2), 71 (2011).
L. Xu and J. Wang: Fenton-like degradation of 2,4-dichlorophenol using Fe3O4 magnetic nanoparticles. Appl. Catal., B 123–124, 117 (2012).
N. Wang, L. Zhu, D. Wang, M. Wang, Z. Lin, and H. Tang: Sono-assisted preparation of highly-efficient peroxidase-like Fe3O4 magnetic nanoparticles for catalytic removal of organic pollutants with H2O2. Ultrason. Sonochem. 17 (3), 526 (2010).
X. Liang, Z. He, Y. Zhong, W. Tan, H. He, P. Yuan, J. Zhu, and J. Zhang: The effect of transition metal substitution on the catalytic activity of magnetite in heterogeneous Fenton reaction: In interfacial view. Colloids Surf., A 435, 28 (2013).
F.F. Peng, Y. Zhang, and N. Gu: Size-dependent peroxidase-like catalytic activity of Fe3O4 nanoparticles. Chin. Chem. Lett. 19 (6), 730 (2008).
Y.F. Shen, J. Tang, Z.H. Nie, Y.D. Wang, Y. Ren, and L. Zuo: Tailoring size and structural distortion of Fe3O4 nanoparticles for the purification of contaminated water. Bioresour. Technol. 100 (18), 4139 (2009).
L. Hou, Q. Zhang, F. Jérôme, D. Duprez, H. Zhang, and S. Royer: Shape-controlled nanostructured magnetite-type materials as highly efficient Fenton catalysts. Appl. Catal., B 144, 739 (2014).
G. Zhang, F. Qie, J. Hou, S. Luo, L. Luo, X. Sun, and T. Tan: One-pot solvothermal method to prepare functionalized Fe3O4 nanoparticles for bioseparation. J. Mater. Res. 27 (7), 1006 (2012).
S. Asuha, B. Suyala, X. Siqintana, and S. Zhao: Direct synthesis of Fe3O4 nanopowder by thermal decomposition of Fe-urea complex and its properties. J. Alloys Compd. 509 (6), 2870 (2011).
A. Prakash, A.V. McCormick, and M.R. Zachariah: Aero-sol−gel synthesis of nanoporous iron-oxide particles: a potential oxidizer for nanoenergetic materials. Chem. Mater. 16 (8), 1466 (2004).
K. Petcharoen and A. Sirivat: Synthesis and characterization of magnetite nanoparticles via the chemical co-precipitation method. Mater. Sci. Eng., B 177 (5), 421 (2012).
L. Li, J. Ding, and J. Xue: A facile green approach for synthesizing monodisperse magnetite nanoparticles. J. Mater. Res. 25 (5), 810 (2010).
A. Yan, X. Liu, G. Qiu, H. Wu, R. Yi, N. Zhang, and J. Xua: Solvothermal synthesis and characterization of size-controlled Fe3O4 nanoparticles. J. Alloys Compd. 458 (1–2), 487 (2008).
H. Meng, Z. Zhang, F. Zhao, T. Qiu, and J. Yang: Orthogonal optimization design for preparation of Fe3O4 nanoparticles via chemical coprecipitation. Appl. Surf. Sci. 280, 679 (2013).
Y. Zhang, W. Shi, D. Feng, H. Ma, Y. Liang, and J. Zuo: Application of rhodamine B thiolactone to fluorescence imaging of Hg2+ in Arabidopsis thaliana. Sens. Actuators, B 153 (1), 261 (2011).
B. Chai, F. Zou, and W. Chen: Facile synthesis of Ag3PO4/C3N4 composites with improved visible light photocatalytic activity. J. Mater. Res. 30 (8), 1128 (2015).
X. Xue, K. Hanna, and N. Deng: Fenton-like oxidation of Rhodamine B in the presence of two types of iron (II, III) oxide. J. Hazard. Mater. 166 (1), 407 (2009).
P.P. Gan and S.F.Y. Li: Efficient removal of rhodamine B using a rice hull-based silica supported iron catalyst by Fenton-like process. Chem. Eng. J. 229, 351 (2013).
X. Wang, Y. Pan, Z. Zhu, and J. Wu: Efficient degradation of rhodamine B using Fe-based metallic glass catalyst by Fenton-like process. Chemosphere 117, 638 (2014).
S. Liu, F. Lu, R. Xing, and J.J. Zhu: Structural effects of Fe3O4 nanocrystals on peroxidase-like activity. Chem. — Eur. J. 17 (2), 620 (2011).
W. Yu, T. Zhang, J. Zhang, X. Qiao, L. Yang, and Y. Liu: The synthesis of octahedral nanoparticles of magnetite. Mater. Lett. 60 (24), 2998 (2006).
S. Yang, H. He, D. Wu, D. Chen, X. Liang, Z. Qin, M. Fan, J. Zhu, and P. Yuan: Decolorization of methylene blue by heterogeneous Fenton reaction using Fe3−xTixO4 (0 < x < 0.78) at neutral pH values. Appl. Catal., B 89 (3–4), 527 (2009).
K. Tao, H. Dou, and K. Sun: Interfacial coprecipitation to prepare magnetite nanoparticles: Concentration and temperature dependence. Colloids Surf., A 320 (1–3), 115 (2008).
G.F. Goya, T.S. Berquó, F.C. Fonseca, and M.P. Morales: Static and dynamic magnetic properties of spherical magnetite nanoparticles. J. Appl. Phys. 94 (5), 3520 (2003).
D. Wan, W. Li, G. Wang, K. Chen, L. Lu, and Q. Hu: Adsorption and heterogeneous degradation of rhodamine B on the surface of magnetic bentonite material. Appl. Surf. Sci. 349, 988 (2015).
T. Yamashita and P. Hayes: Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials. Appl. Surf. Sci. 254 (8), 2441 (2008).
ACKNOWLEDGMENTS
This project was supported in part by the Natural Science Foundation of Hubei Province (2014CFB810), Specialized Research Fund for the Doctoral Program of Higher Education of China (20114219110002) and Science and Technology Project (Major) of Jiangxi Province (20152ACG70003).
Author information
Authors and Affiliations
Corresponding author
Supplementary data for
Rights and permissions
About this article

Cite this article
Wan, D., Li, W., Wang, G. et al. Size-controllable synthesis of Fe3O4 nanoparticles through oxidation–precipitation method as heterogeneous Fenton catalyst. Journal of Materials Research 31, 2608–2616 (2016). https://doi.org/10.1557/jmr.2016.285
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/jmr.2016.285