
Abstract
Tungsten oxide (WO3−x) nanomaterials with controlled morphology and composition were fabricated by thermal evaporation of WO3 and S powders at different temperatures in a vacuum tube furnace. At 850 °C the obtained green particle is still of the same monoclinic WO3 phase as that of the starting powder. At a temperature between 900 and 1100 °C, the resultant dark-blue products are particle-like clusters composed of numerous monoclinic WO2.90 short nanorods, but the clusters became looser and the nanorods grew somewhat longer as the temperature increased. At a temperature between 1150 and 1250 °C, elongated and thoroughly separate purple-red monoclinic W18O49 nanorods were obtained. The growth of the prepared WO3−x nanomaterials was controlled by a gas–solid mechanism. Their photocatalytic degradation on organic contaminants was evaluated by decomposing methylene blue (MB) in aqueous phase under sunlight, in which WO3 particles presented higher photocatalytic activity than its oxygen-deficient counterparts, WO2.90 and W18O49. But the W18O49 nanorods had higher adsorption ability to MB in all the samples.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
I. INTRODUCTION
As a kind of popular metal oxide semiconductor, nanosized tungsten oxides are of great interest due to their outstanding chemical activity, gas sensitivity, electrochromic property, and electromagnetic wave absorption properties.1–3 Up to now, various tungsten oxides have been extensively explored for industrial catalysts,4–7 gas sensors,8 electrochromic materials,9,10 military stealth materials,3 and so on, in which tungsten oxides as photocatalysts have gained intensive attention from the researchers.
As is well known, in the field of photocatalysts, TiO2 has been the best and most widely investigated one in the past 10 years.11 However, due to its wide band-gap of about 3.2 eV, it can functionalize only under ultraviolet (UV) light. As a result, it is difficult for TiO2 photocatalyst to utilize solar energy, the most important and abundant natural light energy that contains only about 4% UV light. To address this issue, new generation of photocatalysts has been intensively developed. On the one hand, many methods have been proposed to modify TiO2, including the doping of anionic or cationic impurity, dye sensitization, construction of surface heterojunctions, the coupling of TiO2 with smaller band-gap semiconductors, increase in the accessible surface areas, and usage of surface F effects and exposure of highly reactive facets.11–14 On the other hand, many non-TiO2 based photocatalysts have been developed. For example, g-C3N4 may be a good visible photocatalyst, because it has a relatively narrow band-gap of about 2.7 eV together with other merits like low cost and no toxicity. However, due to the quick recombination of its photogenerated electron–hole pairs, it still has limited visible photocatalytic efficiency, and this issue might be solved by the fabrication and nanostructure design of pristine g-C3N4, band-gap engineering through atomic-level doping and molecular-level modification, and the preparation of g-C3N4-based semiconductor composites.15,16 Meanwhile, compared with the above photocatalysts, WO3, with a band-gap of 2.8 eV, can also absorb visible light to a large extent besides UV one, and is of high stability but no toxicity, being the second most investigated semiconductor photocatalyst and exhibiting enormous potential in applying solar energy for the splitting of water into hydrogen and oxygen, decomposing of hazardous organic contaminants in polluted water and hazy atmosphere, and so forth.5–7
To date, most of the studies in the literature have focused on the effects of experimental conditions on the photocatalytic performance of tungsten oxides, such as the intensity and spectrum of the irradiation lamp,17–19 pH (potential of hydrogen) value,20,21 temperature,22 concentration,19 and the presence of oxidizing agents20,22 or inorganic ions.20,23,24 However, more and more investigations have revealed that the intrinsic properties of materials composition, morphology, size, and crystal structure together with different modification engineering strategies would present important synergistic effects on their photocatalysis performance.11,25 For example, it was found that nanoscaled tungsten oxide photocatalysts with controlled morphology possessed enhanced photocatalytic activities due to their high purity, novel morphology, and large specific surface area.4,7 More specially, there has been a great potential to manipulate the composition of tungsten oxides, and various WO3−x crystals with a variety of oxygen-deficient stoichiometries, such as W18O49, W5O14, W24O68, and W20O58, have been reported.7,26 All these results inspire us to prepare nanosized tungsten oxides of various compositions and investigate their potential for photocatalysis.
To prepare samples with excellent performance, a number of methods have been developed to synthesize various tungsten oxide nanomaterials, for example, thermal decomposition,27 sol–gel processing,28 hydrothermal method,29 microemulsion method,30 and so on. Compared with these methods, thermal evaporation is more convenient, controllable, and capable of mass-production.31 So, in this work, we propose a simple, fast, clean, and energetically efficient thermal evaporation method for the preparation of WO3−x nanocrystalline materials in large scale, in which only WO3 and S powders are used as the starting materials. Simply by adjusting the evaporation temperature, WO3−x nanorods with controlled morphology and composition were obtained for the first time. And their photocatalytic degradations on organic contaminants were systematically investigated by decomposing the model compound methylene blue (MB) in aqueous phase.
II. EXPERIMENTAL PROCEDURE
A. Materials preparation
The applied WO3 (Sinopharm Group Co. Ltd, Beijing, China) and elemental S (Aladdin Industrial Corporation, Shanghai, China) were commercially bought raw powders of analytical grade and directly used without further purification. The nanocrystalline WO3−x samples were prepared by using an experimental vacuum, horizontal alumina tube furnace (SJG-16, Luoyang Shenjia Kiln Co. Ltd., Henan, China).
In a typical process, 1 g WO3 and 1 g S powders were loaded separately in two alumina boats. The WO3-containing boat was placed at the heating center of the furnace, while another one with S powder was located on the upstream of working gas flow in the furnace, 10 cm away from the WO3 powder. Before heating, the alumina tube was evacuated and flushed repeatedly with high-purity N2 gas so as to eliminate the remnant gases in the tube. Then, the furnace was heated up in 1.5 h from room temperature to a designed heating temperature (800–1300 °C in a step of 50 °C) and held there for 1 h. After that, the furnace was cooled down naturally to room temperature simply by shutting down the electricity of the heating system of the furnace. Throughout the whole heating process, the N2 gas flow inside the alumina tube was maintained at 200 sccm. Finally, powder-like products with different colors could be obtained in the alumina boats originally loaded with WO3 raw powder.
B. Materials characterization
The phase composition of the products was identified by X-ray diffraction (XRD; Bruker D8 diffractometer, Bruker Corporation, Kalsruhe, Germany) with Cu Kα radiation (λ = 1.5418 Å). The XRD scanning range was from 10 to 60° and the speed was 6°/min. Their morphologies and crystallographic structures were examined by field emission scanning electron microscopy (FE-SEM; Hitachi S4800, Hitachi Corp., Tokyo, Japan), transmission electron microscopy (TEM; Tecnai G2 F30 U-TWIN, FEI Company, Hillsboro, Oregon), high-resolution TEM (HRTEM), and selected area electron diffraction (SAED). The UV–vis absorption spectra of solid samples were recorded on a Cary 5000 UV–vis spectrometer (Varian, Salt Lake City, Utah) equipped with a Φ110 mm integrating sphere for solid-phase characterization. And their photoluminescence (PL) spectra were collected at room temperature on a Cary Eclipse fluorescence spectrophotometer (Agilent, Palo Alto, California) using an Ar lamp as the excitation light source at excitation an wave length of 325 nm. Specific surface area was obtained by the Brunauer–Emmett–Teller (BET) N2 adsorption method using a Micromeritics ASAP2020 surface analyzer (China National Pharmaceutical Group Corp., Shanghai, China).
C. Photocatalytic degradation on organic contaminants
The photocatalytic degradation on organic contaminants by the as-synthesized WO3−x samples was evaluated through decomposing the commonly applied model compound MB under 500 W xenon lamp that can generate light as sunlight. The tests were performed in a cylindrical, thermostated (30 °C) and inner lamp type photochemical reactor system (XPA-7, Nanjing Xujiang Mechanical & Electrical Factory, Nanjing, China) under continuous magnetic stirring. In a typical photocatalytic degradation test, 50 mL MB (5 mg/L) aqueous solution was decomposed by 50 mg WO3−x sample in the test tube. During the photocatalytic degradation, the reaction mixture was first stirred in the dark for 60 min to reach the adsorption equilibrium, and then the irradiation lamp was switched on. Throughout the whole tests, after every 20 min reaction, 2–3 mL supernatant sample was collected from the mixture into a small tube. Then the collected supernatant sample was centrifuged for 8 min using HC-2064 High-Speed Centrifuge (Anhui Zhogke Scientific Instrument Co. Ltd., Anhui, China) with a rotation speed of 9000 rpm. After centrifuging, the prepared clear solution from the small tube was analyzed by a 722S Visible Spectrophotometer (Shanghai Lengguang Technology Co. Ltd., Shanghai, China), and the degradation of MB was determined at the wave length of its maximum absorption (664 nm). After the analysis, the clear solution with the sediment during centrifuging was poured back into the test tube. Because the absorbance of MB depends linearly on its concentration according to the Lambert–Beer law, the measured concentration of MB in the clear solution can represent that of the organic contaminant without photocatalytic degradation.
D. Active species trapping experiments
To investigate the active species generated in photocatalytic process, for each photodegradation test on MB by the selected tungsten oxides, 1 mmol tertbutyl alcohol (TBA), disodium ethylenediaminetetraacetate (EDTA), and parabenzoquinone (BQ) were also added into the reaction system, respectively.16,32,33 And all the other experimental parameters during the tests were kept as those of the above photodegradation experiments.
III. RESULTS AND DISCUSSION
A. Composition, structure, and morphology of WO3−x nanomaterials
Figure 1 shows the XRD patterns of typical products fabricated at different temperatures. For comparison, the XRD pattern of the starting WO3 raw powder is also presented. Under the designed conditions, at 850 °C (please also see Fig. S1 in the Supplementary Materials), the obtained sample is still of the same monoclinic WO3 phase as that of the starting powder (JCPDS No. 43-1035). At a temperature between 900 and 1100 °C (also see Fig. S2), we could obtain powders of absolute monoclinic phase of WO2.90. The diffraction patterns of all the samples match perfectly with the standard card JCPDS no. 05-0386 of monoclinic phase of WO2.90. At a temperature between 1150 and 1250 °C (also see Fig. S3), the resultant samples consist of mainly monoclinic W18O49 (JCPDS No. 05-0393) with a small amount of WS2 (JCPDS No. 08-0237). However, at 1300 °C (see Fig. S4), the dominant phase of the synthesized samples is WS2 already (JCPDS No. 08-0237).

XRD patterns of typical products fabricated at different temperatures. For comparison, the XRD pattern of the starting WO3 raw powder was also presented.
Figure 2 presents some SEM images of the samples, in which Fig. 2(a) shows the one of the applied starting WO3 raw powder, and Figs. 2(b)–2(f) exhibit those of typical products prepared at different temperatures. From Fig. 2(a), we can see that the light-green raw powder (also see Fig. S1) consists of many granular particles with various sizes from 10 to 30 µm in diameter. Figure 2(b) shows the SEM image of the sample prepared at 850 °C. From this image, it could be seen that the obtained green powder (also see Fig. S1) still has the same morphology as that of the starting WO3 raw powder. After comparing with their XRD results (also see Fig. S1), it can be inferred that at a temperature ≤850 °C, the prepared samples possess the same composition, structure, and morphology (round monoclinic WO3 particle). The morphology of the powder-like product prepared at 950 °C is displayed in Fig. 2(c). From this image, it seems that the sample is still of the same structure of granular particles as shown in Figs. 2(a) and 2(b), but with a closer view, it could be found that the particles are clusters of many short-length nanorods. The image of the powder-like product prepared at a higher temperature of 1050 °C [Fig. 2(d)] reveals similar particle-like clusters as shown in Fig. 2(c) but of many longer nanorods, while some nanorods are even separated from the particles. In combination with the results as shown in Figs. 1 and S2, it can be seen that at a temperature between 900 and 1100 °C, the obtained dark-blue products are particle-like clusters composed of numerous monoclinic WO2.90 nanorods, but the clusters may become looser and the nanorods grow somewhat longer as the temperature increases. At a temperature between 1150 and 1250 °C, all the obtained purple-red powder-like products are composed of thoroughly separate nanorods with almost entirely straight and uniform geometry (see Figs. 2(e), 2(f), and S3). With a rough evaluation, the diameter of the nanorods is in the range of 200–500 nm, and the length of them is about several to dozens of microns. However, at 1300 °C, the resultant gray powder-like products virtually consist of plate-like nanostructures already, which are the typical structure of nanosized WS2 and can be also confirmed by its XRD pattern (also see Fig. S4).

SEM images of the starting raw powder (a), and typical products prepared at different temperatures: (b) 850, (c) 950, (d) 1050, (e) 1150, and (f) 1250 °C.
To further examine the morphology and structure of the prepared products, TEM, HRTEM, and SAED characterizations were carried out. Typical results are shown in Fig. 3, which were taken from the selected samples prepared at 850, 1050, and 1150 °C, respectively. Their TEM images as shown in Figs. 3(a)–3(c) further confirm the dependence of the geometry of the WO3−x samples on the evaporation temperature. At 850 °C, the prepared sample consists of particles with a diameter of 0.2–2 µm. At 1050 °C, the prepared nanorods possess a diameter of 1–2 µm. And in the sample prepared at 1150 °C, the diameter of the nanorods is only 200–500 nm. All these results are consistent with the SEM observations. Moreover, from the HRTEM images [see Figs. 3(d)–3(f)] and SAED patterns [see the insets in Figs. 3(d)–3(f)], it can be seen that all the selected products are well crystalline, and the growth of the WO3 particles prepared at 850 °C is along the (020) direction, and those of the nanorods prepared at 1050 and 1150 °C are along the (010) and (103) planes, respectively. All these results are basically in accordance with the results recorded by XRD.

(a, b, and c) TEM and (d, e, and f) HRTEM images of typical WO3 particles prepared at 850 °C, WO2.90 nanorods prepared at 1050 °C, and W18O49 nanorods prepared at 1150 °C, respectively. The insets represent their corresponding SAED patterns.
B. Formation mechanism of WO3−x nanomaterials
From the above results, it can be concluded that the temperature of thermal evaporation affects not only the composition of the as-prepared WO3−x nanostructures, but also their morphology. To elucidate the formation mechanism of the present nanostructures, the chemical reactions that happened were first discussed. Considering the starting powders (evaporation sources) applied in this study, it was suggested that as the temperature increased, WO3 and S started to sublimate from the alumina boats separately. When they met with each other, they would first react in the form of
during which tungsten oxide nanostructures with different compositions would be generated at different temperatures, because with increasing degree of oxygen deficiency in the products, the reaction (an endothermic one) would need more heat. When the product is WO2.90, the calculated standard enthalpy of formation for the reaction is 8.004 kJ/mol. But for the product W18O49, it would be 20.20 kJ/mol. Thus the latter reaction requires higher temperature. In a word, with increasing degree of oxygen deficiency in the products, the reaction would need higher temperature. However, at a temperature higher than 1300 °C, the following reaction would be easier to happen,
because it needs much more heat than Reaction (1). In such case, WS2 nanostructures would be obtained. As for the by-product SO2, it was taken away by the carrier gas N2.
To explain the growth process of the present nanorods, three kinds of the already-known mechanisms can be taken into account: vapor–liquid–solid (VLS),34 solid–liquid–solid (SLS),35 and vapor–solid (VS).36 The growth of the present nanorods may not follow a VLS mechanism, because in VLS mode there should exist a clear sign that small spherical droplets appear on the top of nanostructures. However, after careful searching, we could not find such phenomenon on the present tungsten oxide nanorods. Moreover, the growth of the present nanorods would be difficult to follow a SLS mechanism, because the nanorods can be formed only under the action of S vapor (also see Fig. S5), but the applied starting raw powders were loaded separately in two alumina boats, which should be transferred through a gas way for their reaction with each other. So we supposed that the growth of the present tungsten oxide nanorods was controlled by a VS mechanism. In this mode, at an appropriate temperature, the active atmosphere will be adsorbed on the surface of small WO3 particles to form a crystal nucleus. The formed crystal nucleus would continue to grow or decompose with thermal fluctuation in the system. When a certain critical condition is reached, the crystal nucleus will be stable and grow promptly along its preferential growth direction, eventually generating nanostructures. In the present experiment, when the active atmosphere (S vapor) meets WO3 particles, the S vapor will be adsorbed on the surface of WO3 particles to promote WO3 forming a crystal nucleus. When the temperature is less than 850 °C, the formed crystal nucleus would rapidly decompose (not stable), during which the crystalline nucleus would not finally grow. So the final samples are still of particles as the starting raw powder. When the temperature is between 900 and 1100 °C, the formed crystal nucleus is stable, but its formation speed is much higher than that of growth in a specific direction, thus resulting in products with numerous short nanorods. During this stage, the morphology of the nanorods is controlled by the formation of the crystal nucleus. At a temperature between 1150 and 1250 °C, the formed crystal nucleus is also stable, but its growth speed is higher than that of formation. Therefore, the crystal nucleus would grow rapidly along its preferential direction, eventually generating elongated nanorods. That is, during this stage, the morphology of the nanorods is controlled by the growth of the crystal nucleus.
C. Optical properties of WO3−x nanomaterials
To understand the photocatalytic performance of the prepared WO3−x samples, their UV–vis absorption and emission spectra were investigated by using UV–vis and PL spectroscopy. For comparison, the corresponding spectra of the starting WO3 raw powder were also examined.
The recorded UV–vis absorption spectra as shown in Fig. 4(a) reveal that the starting monoclinic WO3 raw powder can only absorb light until approximately 500 nm, while the monoclinic oxygen-deficient WO3−x samples fabricated at different temperatures can have a strong absorption in the whole UV–vis range (200–800 nm). The dark color (absorption above 500 nm) of the oxygen-deficient WO3−x samples is owing to the presence of larger amounts of reduced (W4+ and W5+) atoms as well as W6+, which introduced new discrete, quite deep energy levels into the band-gap.7 However, although the XRD analysis displayed that the sample prepared at 850 °C possesses the same composition and structure as the starting WO3 raw powder, it also has a relatively strong absorption in the whole UV–vis range though with decreased intensity between 400 and 500 nm. This result reveals that the WO3 sample prepared at 850 °C might also have some reduced (W4+ and W5+) atoms in the crystals (thus new discrete, quite deep energy levels into the band-gap), due to the small portion of WO3 reduced by S vapor at a relatively low temperature (lower than 850 °C in this study). But this small portion of reductive WO3−x cannot be detected by XRD in the detection limit. This can thus explain the color difference between the starting WO3 raw powder and samples prepared at 800–850 °C as shown in Fig. S1.

(a) The UV–vis absorption and (b) PL spectra of the starting WO3 raw powder and samples prepared at different temperatures.
The Kubelka–Munk formula was used to calculate the band-gap of the samples from the UV–vis spectra. The band-gap of the starting WO3 raw powder is 2.75 eV, while those of the samples fabricated at 850, 950, 1050, 1150, and 1250 °C are 2.51, 2.22, 2.20, 2.1, and 1.84 eV, respectively. In combination with their compositions presented above, it can be concluded that the band-gap of tungsten oxides decreases with the degree of oxygen deficiency in the samples. On the one hand, due to the stronger absorption in the visible range, the WO3−x samples could be better photocatalysts for the degradation on organic contaminants than the starting WO3 raw powder. On the other hand, the new, quite deep energy levels in their band-gaps might act as recombination centers,37–39 which could lower the photocatalytic efficiency of the WO3−x samples. In addition, the increased conductance of the reductive WO3−x samples, owing to that electrons could easily hop between the W atoms of different oxidation states,26 could result in easier transport and thus recombination of electrons and holes,7 which could also lower their photocatalytic efficiency.
The recorded PL spectra are shown in Fig. 4(b). According to this figure, all the products prepared at different temperatures have similar emission peak systems with the starting WO3 raw powder, in which the strongest emission peak at about 400 nm is close to the UV range. However, the oxygen-deficient WO3−x samples have significantly lower luminescence intensity than the starting WO3 raw powder in the whole spectrum, because the excitation light could induce polaron transitions between W4+, W5+, and W6+ atoms in the reductive WO3−x samples,40 implying that a much weak light could cause the WO3−x samples to generate electrons and holes to participate in the photocatalytic reaction.
D. Photocatalytic degradation on MB
The photocatalytic degradation on organic contaminants by the as-synthesized WO3−x samples was evaluated through decomposing the commonly applied model compound MB on comparison with that by the starting WO3 raw powder. The decolorization effects on MB in aqueous solution by the starting WO3 raw powder and samples prepared at different temperatures during the whole process including the adsorption in the dark and degradation under irradiation are illustrated in Fig. 5(a). And their corresponding −ln(C/C0) versus irradiation time plots during only the irradiation process are shown in Fig. 5(b).

(a) Decolorization effects on MB in aqueous solution by the starting WO3 raw powder and samples prepared at different temperatures during the whole process including the adsorption in the dark and degradation under irradiation, and (b) their corresponding −ln (C/C0) versus irradiation time plots during only the irradiation process.
As seen from Fig. 5(a), after adsorption in the dark for 60 min to the adsorption–desorption equilibrium, the elongated W18O49 nanorods presented the greatest decolorization effects on MB among all the samples. By the typical W18O49 nanorods synthesized at 1150 and 1250 °C, after 60 min of dark adsorption, the concentrations of MB were decreased to 77.2 and 71.7 wt%, respectively, much lower than those of the other samples. To explain this phenomenon, the BET surface areas of the present samples were measured. Figure 6 displays the N2 adsorption–desorption isotherms of the starting WO3 raw powder, typical WO3−x samples prepared at different temperatures, WO2.90 (1050 °C) and W18O49 (1150 °C). It can be seen from this figure that the isotherms of the three samples are characteristic of a type IV isotherm with a type H3 hysteresis loop, indicating that all of them have mesoporous structures.41 However, the BET surface areas of the WO3, WO2.90, and W18O49 samples were determined to be approximately 1.87, 0.46, and 0.42 m2/g, respectively, while the volume of the adsorbed N2 by them were 2.23, 1.5, and 5.2 cm3/g under the relative pressure of 1.0. The abnormally high volume of the adsorbed N2 by the as-synthesized W18O49 nanorods should be attributed to the vacant surface sites enhanced adsorbability in the sample,41 which also has a substantial contribution to the adsorption of N2 by the present WO2.90 sample. This is why the W18O49 nanorods could present the highest adsorption decolorization effect on MB as presented in Fig. 5(a).

N2 adsorption–desorption isotherms of the starting WO3 raw powder and typical WO3−x samples prepared at different temperatures: WO2.90 (1050 °C) and W18O49 (1150 °C).
However, when considering the whole decolorization stage, including the 60 min of dark adsorption and 240 min of sunlight irradiation, the starting WO3 raw powder presented the best total decolorization effect on MB. After test, the measured residual MB concentration could drop down to 25.5 wt%. And the WO3 particles prepared at 850 °C even have the second high activity among all the catalysts, by which the recorded residual MB concentration would decrease to 47.2 wt%. This is because these two catalysts have higher ability of decomposing (oxidizing) MB under sunlight due to their higher chemical state of W atoms: almost completely W6+ ions in the starting WO3 raw powder and very little of W4+ and W5+ ions in the WO3 particles prepared at 850 °C. Correspondingly, for the same reason, the WO2.90 short nanorods (here prepared at 950 and 1050 °C) and elongated W18O49 nanorods (prepared at 1150 and 1250 °C) displayed lower activity of decomposing MB among the catalysts owing to the presence of a larger amount of W4+ and W5+ ions in the compounds. After the total decolorization tests by them, the measured residual MB concentration would drop down to 59.7, 60.1, 58.8, and 54.9 wt%, respectively.
Through their corresponding plots of −ln(C/C0) versus irradiation time as illustrated in Fig. 4(b), the rate constant k values of the photocatalytic reactions with the samples can be calculated. The rate constant of the photodegradation reaction on MB with the starting WO3 raw powder is 0.00512/min, and those with the catalyst samples fabricated at 850, 950, 1050, 1150, and 1250 °C are 0.00252, 0.00148, 0.00146, 0.00114, and 0.00111/min, respectively. This result confirms that with increasing degree of oxygen deficiency, the photodegradation (oxidizing) ability on MB by the WO3−x catalysts under sunlight would decrease. The reason for this phenomenon may be that the new, quite deep energy levels in the band-gaps of WO3−x would act as the recombination centers of photogenerated electrons or holes, and the reductive WO3−x samples, owing to that electrons could easily hop between the W atoms of different oxidation states, could result in easier transport and thus recombination of electrons and holes,7 finally lowering the photocatalytic efficiency of the degradation on MB by the WO3−x catalysts, which can been seen from the UV–vis and PL spectra.
To detect the active species generated in photocatalytic process, under the same conditions for each photodegradation test on MB by the selected tungsten oxides, 1 mmol TBA (a scavenger of hydroxyl radical, ∙OH), EDTA (a quencher of hole, h+), and BQ (a quencher of superoxide radical, ∙O2−) were also added into the reaction system, respectively.16,32,33Fig. 7 shows the photodegradation effects of MB over the starting WO3 raw powder, and typical WO3−x samples prepared at different temperatures, WO2.90 (1050 °C) and W18O49 (1150 °C), in the presence of TBA or EDTA. To determine the degradation ability of EDTA, TBA, and BQ trapping agents on MB, comparative experiments in the absence of catalysts were also conducted (see Fig. 7 and S6). From Fig. 7 and S6, it was found that under the same, designed condition, after photodegradation reaction of 240 min, the decolorized MB by pure TBA was about 12%, that by pure EDTA was up to a quite higher value of about 48%, and that by pure BQ reached at an amazing value of 80%. However, for the photodegradation of MB over WO3, WO2.90, and W18O49 in the presence of TBA scavenger, it was found that the photocatalytic activity of the WO3 sample was largely suppressed by the addition of TBA and the inhibition efficiencies for the degradation of MB is about 67.6% [see Fig. 7(a)]. Thus, it could be inferred that ∙OH serves as the main active species for the photodegradation of MB over WO3 under sunlight irradiation. On the other hand, the photodegradations of MB over the WO2.90 and W18O49 samples were almost not affected by adding TBA as a quencher [see Fig. 7(a)]. Furthermore, the photodegradation effects of MB over the WO3, WO2.90, and W18O49 samples in the presence of EDTA could be improved to a certain extent [see Fig. 7(b)], indicating that the active species during the photodegradation on MB by the present tungsten oxides are not h+. For the WO3 sample, the slight enhancement of photodegradation effect on MB may be derived from the contribution of EDTA, because EDTA alone could present a high visible light degradation efficiency of about 48% on MB. However, for the WO2.90 and W18O49 nanorods, the photodegradation effect of MB would be enhanced dramatically, much higher than that in case of WO3. For this phenomenon, in addition to the contribution of EDTA, the added EDTA might capture a large number of the photogenerated h+, which would suppress the recombination of h+ and e− at the deep level, leading to the improvement of the yield of photogenerated e−. In Ref. 41, Qian et al. proposed that the active species in WO3−x materials were ∙O2− during the photogradation of MB. However, because the added BQ itself is a very high efficient catalyst on the photodegradation of MB (see Fig. S6), the active species cannot be determined from our experiments on the photogradation of MB by the present WO2.9 and W18O49 samples together with BQ. On the basis of the result proposed by Qian et al., after the addition of EDTA into WO3−x, due to the increased yield of photogenerated e−, more ∙O2− with strong oxidation ability might be generated through the reduction of oxygen by the e− of strong reduction ability, finally leading to improved efficiency of photodegradation of MB.

Photodegradation effects of MB over the starting WO3 raw powder, and typical WO3−x samples prepared at different temperatures, WO2.90 (1050 °C), and W18O49 (1150 °C), in the presence of TBA (a) or EDTA (b).
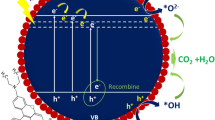
Accordingly, the possible photocatalytic mechanism of WO3−x was proposed in Fig. 8. The low photocatalytic activity of WO3−x should be ascribed to the effect of oxygen vacancies. As shown in Fig. 4(a), the presence of oxygen vacancies could effectively result in a blue-shift on the optical absorption edge, indicating that some new energy levels were formed on the bottom of the conduction band. In this case, the electron was more excited from valence band of WO3−x to arrive at oxygen vacancy level. Because W4+, W5+, or W6+ atom can serve as electron scavenger and ∙O2− producer in the following steps:
and
upon visible light irradiation, photogenerated e− could be trapped by W6+ or W5+, and the W6+ or W5+ would be reduced to W4+. Then, the W4+ could be oxidized back to W6+ or W5+ by the adsorbed oxygen in this system. Meanwhile, O2 adsorbed on the surface of the photocatalyst can react with W4+ and e− to generate ∙O2−. Therefore, the presence of the W4+, W5+, or W6+ atom can result in the generation of ∙O2− for the degradation of the dye molecules. However, excessive W species might cover the active sites or act as a recombination center of WO3−x, which would reduce the separation efficiency of charge carriers, finally reducing the photodegradation activity of WO3−x on MB.

Schematic diagram of the photodegradation mechanism of MB over WO3−x samples under light irradiation.
In a word, when all the factors are taken into account, under the same conditions, the total decolorization effect on MB under sunlight by the present tungsten oxide catalysts can be ranked as: WO3 particles > elongated W18O49 nanorods > WO2.90 short nanorods.
IV. CONCLUSIONS
-
(1)
By only using WO3 and S powders as the evaporation sources, tungsten oxide (WO3−x) nanostructures with controlled morphology and composition were successfully fabricated at different temperatures in a vacuum tube furnace. At 850 °C the obtained green particle is still of the same monoclinic WO3 phase as that of the starting powder. At a temperature between 900 and 1100 °C, the resultant dark-blue products are particle-like clusters composed of numerous monoclinic WO2.90 short nanorods, but the clusters become looser and the nanorods grow somewhat longer as the temperature increases. At a temperature between 1150 and 1250 °C, elongated and thoroughly separate purple-red monoclinic W18O49 nanorods were obtained. And the growth of the obtained WO3−x nanomaterials was controlled by a gas–solid mechanism.
-
(2)
The starting WO3 raw powder can absorb light up to 500 nm in the visible region, while the WO3−x samples had strong absorption in the whole UV–vis range. And their band-gap decreases with the degree of oxygen deficiency in the samples. All the tungsten oxides have similar emission peak systems, but the oxygen-deficient WO3−x samples have significantly lower luminescence intensity than the starting WO3 raw powder in the whole spectrum.
-
(3)
Compared with the oxygen-deficient counterparts WO2.90 and W18O49, WO3 particles presented stronger photocatalytic degradation on MB under sunlight. But the elongated W18O49 nanorods have the highest adsorption ability to MB in all the tungsten oxide samples due to their high content of vacant surface sites.
References
M. Boulova, A. Gaskov, and G. Lucazeau: Tungsten oxide reactivity versus CH4, CO, and NO2 molecules studied by Raman spectroscopy. Sens. Actuators, B 81, 99 (2001).
J.L. Solis, S. Saukko, L. Kish, C.G. Granqvist, and V. Lantto: Semiconductor gas sensors based on nanostructured tungsten oxide. Thin Solid Films 391, 255 (2001).
J. Polleux, N. Pinna, M. Antonietti, and M. Niederberger: Growth and assembly of crystalline tungsten oxide nanostructures assisted by bioligation. J. Am. Chem. Soc. 127, 15595 (2005).
G.C. Xi, J.H. Ye, Q. Ma, N. Su, H. Bai, and C. Wang: In situ growth of metal particles on 3D urchin-like WO3 nanostructures. J. Am. Chem. Soc. 134, 6508 (2012).
B. Rausch, M.D. Symes, G. Chisholm, and L. Cronin: Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting. Science 345, 1326 (2014).
J. Kim, C.W. Lee, and W. Choi: Platinized WO3 as an environmental photocatalyst that generates OH radicals under visible light. Environ. Sci. Technol. 44, 6849 (2010).
I.M. Szilagyi, B. Forizs, O. Rosseler, A. Szegedi, P. Nemeth, P. Kiraly, G. Tarkanyi, B. Vajna, K. Varga-Josepovits, K. Laszlo, A.L. Toth, P. Baranyai, and M. Leskela: WO3 photocatalysts: Influence of structure and composition. J. Catal. 294, 119 (2012).
X.L. Li, T.J. Lou, X.M. Sun, and Y.D. Li: Highly sensitive WO3 hollow-sphere gas sensors. Inorg. Chem. 43, 5442 (2004).
H.G. Wei, X.R. Yan, S.J. Wu, Z.P. Luo, S.Y. Wei, and Z.H. Guo: Electropolymerized polyaniline stabilized tungsten oxide nanocomposite films: Electrochromic behavior and electrochemical energy storage. J. Phys. Chem. C 116, 25052 (2012).
W.T. Wu, J.J. Wu, and J.S. Chen: Resistive switching behavior and multiple transmittance states in solution-processed tungsten oxide. ACS Appl. Mater. Interfaces 3, 2616 (2011).
J.Q. Wen, X. Li, W. Liu, Y.P. Fang, J. Xie, and Y.H. Xu: Photocatalysis fundamentals and surface modification of TiO2 nanomaterials. Chin. J. Catal. 36, 2049 (2015).
R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki, and Y. Taga: Visible-light photocatalysis in nitrogen-doped titanium oxides. Science 293, 269 (2001).
L. Liu and X.B. Chen: Titanium dioxide nanomaterials: Self-structural modifications. Chem. Rev. 114, 9890 (2014).
X.B. Chen, L. Liu, and F.Q. Huang: Black titanium dioxide (TiO2) nanomaterials. Chem. Soc. Rev. 44, 1861 (2015).
S.W. Cao, J.X. Low, J.G. Yu, and M. Jaroniec: Polymeric photocatalysts based on graphitic carbon nitride. Adv. Mater. 27, 2150 (2015).
Z.Y. Wang, W. Guan, Y.J. Sun, F. Dong, Y. Zhou, and W.K. Ho: Water-assisted production of honeycomb-like g-C3N4 with ultralong carrier lifetime and outstanding photocatalytic activity. Nanoscale 7, 2471 (2015).
C.M. Foeldvary and L. Wojnarovits: The effect of high-energy radiation on aqueous solution of Acid Red 1 textile dye. Radiat. Phys. Chem. 76, 1485 (2007).
Y.P. Chen, S.Y. Liu, H.Q. Yu, H. Yin, and Q.R. Li: Radiation-induced degradation of methyl orange in aqueous solutions. Chemosphere 72, 532 (2008).
M. Qamar, M.A. Gondal, and Z.H. Yamani: Synthesis of highly active nanocrystalline WO3 and its application in laser-induced photocatalytic removal of a dye from water. Catal. Commun. 10, 1980 (2009).
M.A. Rauf and S.S. Ashraf: Radiation induced degradation of dyes-an overview. J. Hazard. Mater. 166, 6 (2009).
H.B. Fu, C.S. Pan, W.Q. Yao, and Y.F. Zhu: Visible-light-induced degradation of rhodamine B by nanosized Bi2WO6. J. Phys. Chem. B 109, 22432 (2005).
A. Fujishima, X.T. Zhang, and D.A. Tryk: TiO2 photocatalysis and related surface phenomena. Surf. Sci. Rep. 63, 515 (2008).
G.R. Bamwenda, K. Sayama, and H. Arakawa: The effect of selected reaction parameters on the photoproduction of oxygen and hydrogen from a WO3–Fe2+–Fe3+ aqueous suspension. J. Photochem. Photobiol., A 122, 175 (1999).
L. Wan, J. Sheng, H. Chen, and Y. Xu: Different recycle behavior of Cu2+ and Fe3+ ions for phenol photodegradation over TiO2 and WO3. J. Hazard. Mater. 262, 114 (2013).
X. Li, J.G. Yu, J.X. Low, Y.P. Fang, J. Xiao, and X.B. Chen: Engineering heterogeneous semiconductors for solar water splitting. J. Mater. Chem. A 3, 2485 (2015).
I.M. Szilagyi, J. Madarasz, G. Pokol, P. Kiraly, G. Tarkanyi, S. Saukko, J. Mizsei, A.L. Toth, A. Szabo, and K. Varga-Josepovitso: Stability and controlled composition of hexagonal WO3. Chem. Mater. 20, 4116 (2008).
T.J. DeJournett and J.B. Spicer: Photoinduced silver precursor decomposition for particle modification in tungsten oxide-polymer matrix nanocomposites. J. Phys. Chem. C 118, 9820 (2014).
E.K. Papynov, V.Y. Mayorov, M.S. Palamarchuk, and V.A. Avramenko: Peculiarities of formation of phase composition, porous structure, and catalytic properties of tungsten oxide-based macroporous materials fabricated by sol–gel synthesis. Mater. Charact. 88, 42 (2014).
M. Ahmadi, R. Younesi, and M.J.F. Guinel: Synthesis of tungsten oxide nanoparticles using a hydrothermal method at ambient pressure. J. Mater. Res. 29, 1424 (2014).
C.Z. Yang, N.K. van der Laak, K.Y. Chan, and X. Zhang: Microwave-assisted microemulsion synthesis of carbon supported Pt-WO3 nanoparticles as an electrocatalyst for methanol oxidation. Electrochim. Acta 75, 262 (2012).
J.W. Qian, Z.Y. Zhao, Z.G. Shen, G.L. Zhang, Z.J. Peng, and X.L. Fu: A large scale of CuS nano-networks: Catalyst-free morphologically controllable growth and their application as efficient photocatalysts. J. Mater. Res. 30, 3746 (2015).
F.J. Wu, W. Liu, J.L. Qiu, J.Z. Li, W.Y. Zhou, Y.P. Fang, S.T. Zhang, and X. Li: Enhanced photocatalytic degradation and adsorption of methylene blue via TiO2 nanocrystals supported on graphene-like bamboo charcoal. Appl. Surf. Sci. 358, 425 (2015).
H.W. Huang, K. Liu, K. Chen, Y.L. Zhang, Y.H. Zhang, and S.C. Wang: Ce and F comodification on the crystal structure and enhanced photocatalytic activity of Bi2WO6 photocatalyst under visible light irradiation. J. Phys. Chem. C 118, 14379 (2014).
R.S. Wagner and W.C. Ellis: Vapor–liquid–solid mechanism of single crystal growth. Appl. Phys. Lett. 4, 89 (1964).
J.Y. Chen, B.J. Wiley, and Y.N. Xia: One-dimensional nanostructures of metals: Large-scale synthesis and some potential applications. Langmuir 23, 4120 (2007).
T.J. Trentler, K.M. Hickman, S.C. Geol, A.M. Viano, P.C. Gibbons, and W.E. Buhro: Solution–liquid–solid growth of crystalline III–V semiconductors: An analogy to vapor–liquid–solid growth. Science 270, 1791 (1995).
L. Palmisano, V. Augugliaro, A. Sclafani, and M. Schiavello: Activity of chromium-ion-doped titania for the dinitrogen photoreduction to ammonia and for the phenol photodegradation. J. Phys. Chem. 92, 6710 (1988).
J.M. Herrmann, J. Disdier, and P. Pichat: Effect of chromium doping on the electrical and catalytic properties of powder titania under UV and visible illumination. Chem. Phys. Lett. 108, 618 (1984).
O. Carp, C.L. Huisman, and A. Reller: Photoinduced reactivity of titanium dioxide. Prog. Solid State Chem. 32, 33 (2004).
S.H. Lee, H.M. Cheong, C.E. Tracy, A. Mascarenhas, D.K. Benson, and S.K. Deb: Raman spectroscopic studies of electrochromic a-WO3. Electrochim. Acta 44, 3111 (1999).
J.W. Qian, Z.Y. Zhao, Z.G. Shen, G.L. Zhang, Z.J. Peng, and X.L. Fu: Oxide vacancies enhanced visible active photocatalytic W19O55 NMRs via strong adsorption. RSC Adv. 6, 8061 (2016).
ACKNOWLEDGMENTS
The authors would like to thank the financial support for this work from the National Natural Science Foundation of China (Grant Nos. 61274015, 11274052, and 51172030), Excellent Adviser Foundation in China University of Geosciences from the Fundamental Research Funds for the Central Universities, and Fund of State Key Laboratory of Information Photonics and Optical Communications (Beijing University of Posts and Telecommunications).
Author information
Authors and Affiliations
Corresponding authors
Supplementary Material
43578_2016_31081065_MOESM1_ESM.docx
Supplementary Materials: Synthesis of WO3−X nanomaterials with controlled morphology and composition for highly efficient photocatalysis (approximately 33.2 MB)
Rights and permissions
About this article

Cite this article
Shen, Z., Zhao, Z., Qian, J. et al. Synthesis of WO3−x nanomaterials with controlled morphology and composition for highly efficient photocatalysis. Journal of Materials Research 31, 1065–1076 (2016). https://doi.org/10.1557/jmr.2016.106
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/jmr.2016.106