
Abstract
With the aging of the world population, there has been a notable increase in the incidence of Alzheimer disease (AD), the most prevalent neurodegenerative disease affecting the elderly. Several studies have reported a delay in the onset of AD symptoms and age-related cognitive dysfunction upon changes to a healthier lifestyle. These positive adjustments find support in the cognitive reserve hypothesis, which holds that the ability to defer disease inception and protect cognitive performance is related to healthier lifestyle habits such as cognitive and physical activity, social engagement, and sensorial stimulation. These lifestyle habits can be compounded under the umbrella of the environmental enrichment (EE) paradigm. The mechanisms underlying EE’s capacity to modulate disease expression remain unclear. Since ethical and methodological considerations rule out direct analysis of such changes in the human brain, researchers have resorted to animal models to carry out in-depth characterizations of post-EE structural and functional brain modifications using a variety of behavioral, electrophysiological, genetic, biochemical, and biophysical approaches. Moreover, given the shorter lifespan of animals compared to humans, it is possible to address the effects of aging in control and AD models. In this review we analyze and classify EE data from studies using AD murine models and compare the setup variables employed. We also delve into various aspects of neuroplasticity, under the posit that this property is the key mechanistic process underlying the benefits of EE in both animal and human subjects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The brain has a remarkable capacity to reorganize itself at both the structural and functional level in response to intrinsic and extrinsic stimuli, following mechanisms collectively comprised under the umbrella of the term “neural plasticity” or “neuroplasticity” (1). This capacity is not limited to early periods of neurodevelopment and is currently assumed to persist throughout the lifespan of the individual (2). Though anatomo-functional changes during aging may negatively affect this capacity, there is evidence that their detrimental consequences can be attenuated by training via sensorial, cognitive, physical, and social stimuli of the nervous system (3, 4).
EE is mostly studied in animal models and consists of interventions that facilitate sensory, cognitive, motor and social activity (5). The approach has been used to explore the connection between brain structure and function and it has been found that EE can alter the morphology and molecular biology of animal brains by increasing the number and volume of dendritic spines, enhancing neuroplasticity, and delaying memory decline in aged rats (3).
There is growing interest in behavioral interventions that can positively modify the course of cognitive decline or other dysfunctional cognitive conditions, especially in view of the higher prevalence of age-related cognitive impairments in contemporary life. It is expected that the world’s population of people aged 65 and older will reach approximately 2.1 billion by 2050. Since aging is the highest risk factor for dementia, and Alzheimer disease (AD) is the most common form of dementia, the number of people suffering from AD will increase as the global population ages, reaching 115.4 million people by 2050, according to the World Health Organization (6, 7). The outcome of the many disease-modifying prophylactic or therapeutic endeavours has so far not been promising (8, 9). Improving and maintaining a healthy lifestyle during aging is one of the current focuses of research (4, 10). Indeed, an active lifestyle has been proposed as a means to delay the appearance of age-related deficits and promote the healthy aging of the brain, positively affecting brain plasticity (11, 12). Cognitive stimulation, physical activity and frequency of social activities have been shown in various studies to produce beneficial effects, thus encouraging the search for interventions and activities to promote the buildup of cognitive reserve (13, 14). Little is known about the effect of these interventions on human brain plasticity and structure, and in this scenario, animal models provide a useful alternative to investigate the brain mechanisms involved in the development of cognitive reserve triggered by a positively stimulating environment (15). Compared with traditional drug and/or immune therapies, providing a stimulating environment is deemed to create stress-free, beneficial conditions for individuals with AD or other forms of dementia. This approach can improve mood, behavior, and communication as well as enhance cognition, enabling the level of medication to be reduced and thus avoiding the side effects of drug therapy (16). Numerous factors intervene in the aging process, modifying the likelihood of developing dementia, and it is known that the complex interaction between genes and life experiences plays a significant role in AD pathophysiology. Encouraging engagement in stimulating activities is therefore considered nowadays to be a promising strategy for providing neuroprotective functions (11, 17–19).
This review explores environmental factors in relation to AD and critically discusses therapeutic strategies based on the biological and behavioral outcomes of EE interventions in animal models of AD and their possible translation into clinical practice for the treatment of AD in humans. Literature on rodent models of EE in the context of AD during the last decade were searched in PubMed using “Environmental Enrichment”, “Alzheimer Disease”, “Cognitive Reserve” and “Animal Models” as keywords. The variability in EE setups in the retrieved studies is examined and the findings critically discussed.
Alzheimer disease (AD) and environmental factors
Alzheimer Disease (AD) is the most prevalent form of dementia, currently representing around 60% to 70% cases of dementia worldwide (6). Autopsies of AD brain tissue reveal two histological hallmarks: extracellular deposits of the protein β-amyloid (Aβ) and intracellular accumulations of hyperphosphorylated tau protein, i.e., neurofibrillary tangles, mainly in brain regions involved in learning, memory, and emotional processes, like the hippocampus, cerebral cortex and amygdala (7). Both proteins progressively accumulate during the preclinical stage of AD, devoid of symptomatic cognitive decline, which may last up to 20 years (20). Other widely reported additional pathological features of AD (21) include brain atrophy, synaptic disintegration and neuronal loss, as well as dysregulation of certain elements like zinc, iron and copper. Current research focuses on the development of specific AD biomarkers (22) for detection in cerebrospinal fluid or blood assays as an aid to diagnosis, not as diagnostic tools themselves (23).
AD can show heterogeneous cognitive deficits, such as visuospatial, language, or praxis difficulties, accompanied by the relative sparing of memory (24). Initial loss of episodic memory impeding the recollection of recent events in AD patients can develop into progressive cognitive decline along with other symptoms such as wandering, physical aggression and sleep disturbances, among others (25). Psychiatric features such as psychosis, apathy and depression (26, 27) are also quite frequent in AD patients, constituting an important source of distress and lowering their quality of life (28). The growing incidence of AD alongside lack of knowledge about its pathogenesis and pathophysiology makes further research in this area imperative (29). Familial cases of AD, the least prevalent (< 5%) form of the disease, are caused by genetic mutations (30), whereas in the predominant, sporadic, late-onset form of the disease (LOAD), environmental factors have been suggested to play a major role in its etiology, although some genetic factors may also contribute (31). Numerous epidemiological studies have identified education and occupational attainments as environmental factors that may affect the risk of developing AD, the risk diminishing significantly in those cases that include an enriched cognitive, physical, or social environment (4, 32). The most important non-genetic risk factors are age and gender: women are more prone to develop AD, and though this has been attributed to their greater longevity, it has been suggested that there may be factors other than longevity accounting for this propensity, such as reproductive record and pregnancies, but the underlying mechanisms for this higher risk in women remain largely unclear (33).
The hypothesis of a linear cascade initiated by amyloid and leading to dementia, implying direct causation in the pathogenesis of AD, needs to be revised (34, 35). Although much remains unknown, there is evidence that the immune system plays a role in animal models of AD, showing a more pronounced impairment of the neuroimmunoendocrine network in transgenic murine models of AD than in normally aging mice (36, 37). There are conflicting reports in the literature regarding neuroplasticity in human brains and animal models, suggesting that AD pathology decreases neurogenesis and maturation of newborn neurons (38). Most of the studies report reduced neurogenesis in AD pathology, but the reason for this is not completely understood: some data suggest a direct toxic effect of Aβ deposition (33), whereas other factors such as neuroinflammation (36), loss of cholinergic input (39), and altered levels of growth factors, have also been associated with impaired neurogenesis in transgenic AD mice (40).
Evidence in humans suggests that environmental stressors could also increase the risk of AD (Bisht et al., 41) and animal studies demonstrate that stress stimuli and stress hormones -biological proxies of human chronic stress- constitute risk factors for AD (18). Furthermore, increased stress exacerbates Aβ production and deposition in transgenic AD murine models, and several health factors, such as cardiovascular disease, ischemic strokes, type II diabetes and depression, result in increased risk of developing sporadic AD (29).
It should be underlined that the presence of the characteristic histopathological features of AD is not necessarily associated with cognitive impairment or functional deterioration, as normal elderly people may present these features too (17). A classic example is the Nun study, a longitudinal study of 678 catholic nuns between 75 and 107 years of age, carried out by Snowdon at the University of Minnesota. This study collected data on early and mid-life risk factors, annual cognitive and physical function assessment, and postmortem neuropathology, to present cases that resemble healthy aging. The main finding of this investigation was the lack of a direct relationship between disease neuropathology and cognitive function, manifested by the level of resistance to the clinical expression of brain pathology present in those individuals with a healthy lifestyle history (42). The severity of Aβ and tau accumulation has been shown not to be associated with cognitive function; however, other hallmarks like synaptic density appear to be correlated with dementia in postmortem studies, indicating that factors other than Aβ accumulation are associated with cognitive performance, as observed in murine models of AD (38). Moreover, some authors investigated the occurrence of cognitive reserve in a longitudinal study assessing the topography and extent of hypometabolism progression in patients with mild cognitive impairment (MCI), comparing aggressive versus smoldering conversion to AD dementia, and stratifying the groups according to educational level (years of schooling) (43). They found that brain regions associated with a higher metabolic resistance and slower clinical progression (middle and inferior temporal gyri) were different from those regions affected by education-related reserve (posterior cortical areas), suggesting a specific topographic target for assessing the effect of lifestyle enrichment and risk factors associated with AD.
There has been growing interest in studying the environmental risk factors that modify the amyloid cascade and AD progression, leading to interventions for the prevention and treatment of AD symptoms and neuropathology, both in animals and humans (44). Research findings show that different lifestyle factors, including level of education, mental stimulation, social activities, and physical activity, correlate with both the delayed onset and lower incidence of AD, suggesting that there are modifiable environmental factors able to modulate the progression to dementia (12).
The environmental enrichment (EE) paradigm
The concept of EE was originally introduced by Donald Hebb in the 1940’s as he tried to understand the role of experience in brain development. In his famous book, “The Organization of Behavior” (45) he argued that intelligence is not directly correlated with the integrity of brain structure, and that at least part of it is a product of experience. At that time, intelligence was considered an innate trait, unmodifiable by environmental stimuli. Soon it was demonstrated that enrichment had lasting and generalized effects on behavior and learning, and that these changes responded to substantial effects of EE on the structure of the brain; henceforth, interest therefore shifted to developing the idea of how experience shapes brain structure and function (44).
The EE paradigm has mostly been probed in animal models, facilitating motor, sensory and cognitive stimulation, thus providing the means to dissect and explore gene-environment interactions in diverse neurodegenerative diseases, such as AD, Huntington disease, Parkinson disease, fragile X syndrome, Down syndrome and various forms of brain injury (46). EE in animal models involves cages that are larger than standard housing, contain more animals per cage, and are equipped with different interactive objects intended to enhance sensory, cognitive, physical, and social stimulation. The key factor of the EE experimental paradigm is exposure to an enhanced, novel and complex milieu relative to standard housing conditions (3).
The benefits of an enriched environment prompted further interest in studying its effects on behavioral function and brain structure under disease conditions. One encouraging observation was the first report that EE delayed the onset of symptoms in a genetic model of Huntington disease (47), later confirmed by Hockly and coworkers (48) and Spires and coworkers, who further found that Huntington disease-induced downregulation of BDNF and DARPP-32 protein levels is ameliorated by EE, suggesting potential therapeutic targets (49). In the case of animal models of AD, Arendash and coworkers found that EE improved cognition in aged mice, despite the occurrence of β-amyloid deposition in these animals (50). Similarly, EE and in particular its cognitive and social components were found to be of great importance in improving performance in the Tg-SwDi mouse, a transgenic model of cerebral amyloid angiopathy, a vascular pathological condition common to both aging and AD (51). In essence, EE in animal models mimics beneficial lifestyles in humans that promote plasticity for the build-up of cognitive reserve (52, 53). Overall, these studies have shown that EE exposure in these models of pathology can ameliorate cognitive deficits, though some findings provide confusing explanations of this functional improvement and furthermore the molecular mechanisms underlying these positive effects have not been fully elucidated (54). As summarized in Figure 1, the experimental EE model consists of four central elements (46, 52).

Essential components of the EE paradigm
The sensory stimulation component of the EE in animal models consists of elements of different textures and colors. The cognitive component involves stimulation via various tests and the novelty factor, thereby, promoting learning. The physical component comprises wheels, toys, tunnels, boxes, swings, and ladders in the cages that are useful for stimulating the animals’ voluntary exercise. Lastly, the social component results not from external objects but from the use of a sufficient number of individual animals per cage to generate socialization among them (3).
Transgenic murine models expressing some sign or symptom of AD constitute an important resource to characterize the effects of EE on this disease (37). However, the ability of these models to predict clinical outcomes has often been challenged, as multiples issues surround their validity and predictability. Firstly, the biological differences between rodents and humans are by far the most important factor. Secondly, the complex and still obscure etiopathology and heterogeneity of AD make it unlikely that a given model, developed to incorporate a particular aspect of the disease, reflects the entire broad spectrum of (human) AD (55, 56). The researcher must therefore be aware of the limitations of the animal model approach, and select a model in accordance with the question to be addressed (57). The main types of AD mouse models used in EE studies on AD are listed in Table 1.
Mice with amyloid plaque pathology have been intensively studied in most EE studies, showing functional improvements under enriched conditions, though the effects of the latter on the course of the neuropathology are variable. Despite the broad availability of murine models to study AD, these discrepancies suggest that amyloid pathology, even though considered one of the central hallmarks in AD patients, may not be the primary variable that changes in response to stimulating activities in the animal models. In this regard, the complexity of the underlying mechanisms of responsiveness is still far from being understood (66). Overall, the EE paradigm has become one of the most successful general concepts in experimental biology and psychology as it allows researchers to explore the fundamental link between structure and function (37, 44). Revision of the literature points to the lack of a standardized setup model, as analyzed in the next section. Furthermore, large variations in the methodological paradigms employed by different studies are apparent, even though the central variables remain relatively similar.
EE setups
The lack of an agreed protocol for EE in AD has led to a wide variability in set-ups among different research groups and to difficulties in establishing comparisons. One of the many variables that contribute to this issue is the rodent model used. Even though most studies employed transgenic rodents with stable expression of AD-like pathology (e.g., 5xFAD, Tg2576, APP/PS1 and 3xTg mice), some investigations used wild-type mice injected with induced AD-like pathology (36, 39, 66–69). Only a few studies employed different models, e.g., a transgenic model (Tg-SwDI) of cerebral amyloid angiopathy (51), or a model of senescence-accelerated mice (64, 70).
Sex of the animals differ across studies too, even though the majority of enrichment studies used female animals. The reason why most of them prefer to use female rather than male animals is to avoid possible inter- male aggressive behavior (71, 72).
There are also variations in the number of animals per group in EE versus standard housing. While some studies include the same number of animals in enriched environments as in standard environments, thus eluding the stimulation of the social component of the EE paradigm (70, 71, 73–81), other studies include social stimulation in EE housing, with animals ranging from 3 (40, 50, 82–84) to around 20 per cage (31).
The age of the animals at onset of the EE training also varies widely among studies, with some addressing the effects of a stimulating environment commencing at postnatal life and/or infancy (18, 31, 36, 40, 54, 67, 72, 75–79, 84–86), at pre-clinical stages of the disease using young adult mice (29, 37, 39, 53, 64, 66, 68, 80, 83, 87–91), or at later stages, when the disease had already manifested (16, 50, 51, 73, 81, 82, 92). This parameter is worth considering, as enrichment can have a different impact depending on the age of the animals at the beginning of the experiment: if it commences prior to adulthood, there may be additional effects on the developing brain that would not be observed in those whose exposure began later in life (46).
The duration of the EE intervention also differed between studies, ranging from a couple of weeks (91) to over 12 months (93). In sum, the onset of exposure to EE, as well as the age of the animals at the beginning of the experiments, are not consistently unified variables and may account for some of the discrepancies of the results. We consider it important to address the differential capacity of the EE paradigms with explicit consideration of the age of exposure onset and its duration.
Lastly, the material setting was similar in almost all studies. Cages contained different colored objects, nesting material, toys, at least one running wheel for voluntary physical exercise, and other mice serving as social stimuli. Novelty is an essential factor that should be present to stimulate the cognitive component and promote learning (3), and it was present in almost all studies except for one (73) in which there was no periodical rearrangement and substitution of objects in the cages. Further research is required to quantitatively assess the value of each contributing factor in the outcomes reported in EE studies with the ultimate aim of dissecting their effect on the brain of AD mouse models.
EE outcomes in animal model trials of AD
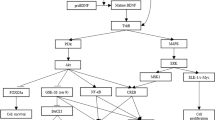
At the anatomic and functional levels, the brains of animals exposed to EE significantly differ from those living in the deprived conditions of standard cages (94). Animal studies in the EE paradigm use two broad experimental measures -biological and cognitive outcomes- via analysis of molecular and/or supra-biological factors and performance in cognitive tasks, respectively (95). In the literature revised here involving specifically rodent model trials of EE in AD, the following outcomes, which can be grouped into five categories (Figure 2), were measured: growth factors and neurogenesis, impact on disease neuropathology, modifications of cholinergic mechanisms, neuroglia, and behavioral factors.

Categories of outcomes in AD animal model trials upon environmental enrichment
Growth factors and adult neurogenesis
The importance of growth factors, such as brain-derived neurotrophic factor (BDNF), nerve-growth factor (NGF), glial cell derived neurotrophic factor (GDNF), and neurotrophin-3 (NT-3) -among others- lies in their role in adult neural plasticity (3). There is evidence, albeit contradictory, that expression of a variety of cellular growth factors is augmented in AD brains upon EE. Some authors found a significant increase in BDNF levels in mice exposed to EE, suggesting that this neurotrophic factor may mediate the beneficial effects of EE (66, 73). In addition to BDNF, increased levels of NGF in the hippocampus of mice exposed to EE has also been reported (16, 54, 96). Hüttenrauch et al. [40] found alterations at the level of gene expression: BDNF mRNA levels changed in the brains of enriched-housed AD mice, reaching the levels found in control wild-type mice.
Several studies have examined adult neurogenesis in AD mouse models, with conflicting outcomes (33). Cotel et al. (79) did not find neurogenesis restored after EE exposure in AD transgenic mice (as found in wild-type mice), results that contradict those reported by Mirochnich et al. (97), who found that EE is able to increase adult neurogenesis at 6 and 18 months of age, and by Jeong et al. (91), who also find neurogenesis, specifically in the dentate gyrus. Other authors also found that hippocampal neurogenesis could be restored after living in an enriched environment (98). Herring and coworkers (86) found that EE was able to improve plasticity after 4 months of exposure, by increasing newborn hippocampal cells and upregulating plasticity associated molecules. Furthermore, higher synaptic densities were observed in the dentate gyrus after combining EE and magnesium intake by Huang et al. (93), along with a behavioral improvement presumably due to the activation of the N-methyl-D-aspartate receptor (NMDAR) pathway.
Impact on disease neuropathology
As mentioned, earlier, the main histopathological hallmarks of AD are Aβ plaques and neurofibrillary tangles. It has been shown that exposure to EE at young ages attenuates amyloid deposition, but not if exposure was performed on 10 months-old animals (99). According to this study, this finding was independent of cognitive performance, as mice exposed to EE at the age of 5 months did produce cognitive benefits, even though their amyloid deposits did not differ from standard-housed transgenic mice. Similarly, Jankowsky and coworkers (75, 100) found that EE rescued deficits in spatial memory despite an increased amyloid burden. Polito et al. (81) reported an amelioration of amyloid deposition after a long (18 months) but not after a short (8 months) exposure to EE. Griñán-Ferré et al. (80) found changes in AD markers after EE exposure: the enrichment paradigm prevented tauopathy and the amyloid cascade in transgenic female mice after 8 weeks of EE training. Zhang et al. (16) also found attenuated amyloid plaque deposition. Interestingly, Maesako et al. (101) found that the increased Aβ deposition resulting from a high-fat diet could be reversed by a 10 week-long EE exposure, even when the high-fat diet was sustained. However, other studies found no difference in Aβ plaque load between EE-type housing and standard housing (31, 37, 82, 84, 88). In pregnant mice, EE and voluntary exercise avoided Aβ pathology and vivified neurogenesis (33).
One study found that even though EE reduced microvascular amyloid deposition in the thalamus compared to the standard-caged mice, socially stimulated mice alone showed even less vascular amyloid pathology compared to fully enriched mice, i.e., those exposed to all the components of the EE simultaneously (51). Herring et al. (77) concluded that EE can counteract AD’s neurovascular dysfunction in transgenic mice by increasing angiogenesis and suggested that these neurovascular changes may be partly responsible for the reduction in Aβ plaques and amyloid angiopathy (although soluble brain and blood soluble Aβ levels remained unchanged in this study). Jeong and coworkers showed that even though EE diminishes the increase in stress-induced tau levels in the hippocampus, it did not alter amyloid plaque deposition or soluble Aβ levels (91), in contrast with the study of Berardi et al. (83), who found that hyperphosphorylated tau was not reduced by EE. Lastly, one study found changes in the regulation of micro-RNA levels relevant to AD pathology upon EE in AD transgenic mice, thereby ameliorating a-synuclein accumulation (78). Some authors (66) found a reduced burden of neurofibrillary tangles after EE, and Dong et al. (70) found that the combined treatment of EE and memantine reduced tau protein aggregation and decreased amyloid precursor protein (APP) levels in the hippocampus. Interestingly, the changes in the levels of APP were not observed in mice exposed to EE alone.
Modifications of cholinergic mechanisms
Cholinergic neurotransmission in cortical and hippocampal brain regions is involved in the adequate functioning of several cognitive processes such as attention, learning and memory (102), and in brain plasticity and homeostasis (103). The cholinergic system comprises cholinergic neurons, whose most important innervation emerges from the basal forebrain, reaching cortical and subcortical regions (7), as well as muscarinic and nicotinic acetylcholine receptors. The former are G-protein coupled receptors involved mainly in autonomic functions, whereas brain nicotinic receptors are ligand-gated ion channels with an important participation in memory and learning processes (104). The normal process of aging involves a gradual loss of cholinergic function, caused by dendritic, synaptic and axonal degeneration, which leads to brain atrophy and the normal cognitive decline observed in elder individuals. In contrast, AD is characterized by a more severe alteration of the cholinergic system, even though the diminution of cholinergic innervation has only been reported in advanced and not preclinical or prodromal stages (7). The integrity of the cholinergic system, especially nicotinic receptors, is considered to be essential for the maintenance of cognitive performance and is a promising therapeutic target for mitigating the symptoms of AD (105, 106).
Despite the importance of the cholinergic system in AD pathophysiology and as a therapeutic target, only three studies involving the EE paradigm consider the cholinergic function and they explore only one aspect of the cholinergic system, namely acetylcholinesterase activity, upon EE interventions. Findings were contradictory: one study found no differences between transgenic mice exposed to EE and controls (39), whereas another study found decreased levels of acetylcholinesterase in hippocampus and cortex in AD model mice reared in an enriched environment (16). Berardi and coworkers (83) reported that EE entirely prevented the loss of the acetylcholine synthesizing enzyme, choline acetyltransferase-positive neurons in the nucleus basalis of Meynert and the medial septum-diagonal band of Broca. Considering its importance in AD etiopathology, modifications to the cholinergic system upon EE should be further explored in future investigations.
Neuroglia
Evidence suggests that neuroinflammation is a significant contributor to AD pathogenesis, and that the brain’s immune system, particularly the microglia, would play a major role in producing/sustaining/reducing inflammation, contributing on the one hand to both neuronal dysfunction and apoptosis and on the other, to reduction of the inflammatory processes. Herring et al. (76), for example, have reported a downregulation of pro-apoptotic caspases, pro-inflammatory and prooxidative mediators upon housing enrichment, as well as an induction of an anti-oxidative defense mechanism. In this context, EE may require the presence of non- neuronal cells, such as microglia, for exerting its benefits. However, though the role of microglia in AD has been extensively studied, the influence of microglia on AD pathobiology in relation to EE has barely been addressed (36). Among the few studies on this subject, Arranz et al. (89, 90) found that EE can reverse the adverse effects of aging on the neuroimmuno-endocrine axis, extending lifespan of transgenic mice upon exposure to EE. This appears to constitute a good strategy for stimulating the immune system in brain, with an overall enhancement of its functions. For example, Cao and coworkers (84) found that the physical component of EE can rescue neuroinflammation and glial activation in transgenic AD mice, counteracting even the detrimental factors of social isolation, i.e., lack of the social component of EE. Xu et al. (36) found that prolonged EE increases microglia density in the hippocampus and results in a prominent neutralization of the neuroinflammation induced by β-amyloid oligomers. It also enhances the immune response of the microglia, showing a protective effect on AD at the level of central nervous system immune response. Changes in microglia were only seen in the hippocampus and not in cortical areas. In line with this, Stozicka et al. (96) found that EE prevented the inflammatory processes promoted by the upregulation of microglia, rescuing functional impairment without altering the levels of tau pathology. Moreover, independently of the mouse model used, Aβ deposition leads to a decline in the number of immature neurons and proliferation, i.e., impaired neurogenesis, which can be circumvented by exposure to EE and voluntary running. This vivifies neurogenesis and rescues apoptosis, suggesting an overall beneficial effect of EE on the survival of neurons. According to Ziegler-Waldkirch et al. (33), this mechanism is most likely driven by the abundance of phagocytic microglia. Apparently, changes in microglia are in part due to running opportunities, pointing to this activity as being responsible for microglial activation. This mechanism could also account for the restoration of impaired neurogenesis in transgenic mouse models. In contrast, one study reported a diminution of microglia upon EE exposure. The discrepancy might be due to the different transgenic lines and age of the animals (87). Another study found no changes in microglia upon EE exposure (37), whereas Stuart et al. (73) found that transgenic mice show an increased number of phagocytic microglia upon EE, in contrast with those from standard housing. The role of microglia in AD has been extensively studied, but studies concerning the effects of EE exposure on microglia are still not conclusive and show contradicting results. Further research with detailed description of the setups is needed to clarify the mechanisms involved.
Only one study on astroglia was found in the literature search (107). These authors reported that EE can modulate and prevent the early pathological alterations induced by astrocytes in the hippocampus of an adult transgenic mouse model of AD and promote morphological changes like those found in healthy mice. More research is required on this topic.
Behavioral factors
According to the reviewed literature, the predominant effect of EE on animals is an improvement in certain behavioral outcomes. EE enhances working memory, with clearer benefits in females (108), although two other studies have not been able to reproduce such an improvement (40, 79). Less contradictory results have been reported regarding spatial learning and memory, with almost unanimous reports on improved performance upon EE (31, 33, 50, 64, 66, 81, 82, 84, 91, 92, 99, 100, 108). Berardi et al. (83) suggested that EE can delay the onset of memory deficits. The exception is a study (40) suggesting that EE intervention might be too mild to counteract fast progression in the early onset familial subtype of AD (EOAD). These authors hold that stimulation must consider the severity and subtype of pathology. Object and social recognition have also been reported to be enhanced upon EE (39, 99). Learning and memory performance, in general, were improved following EE (16, 73, 88, 99), though long-term memory performance in transgenic mice was not as enhanced as in wild-type mice subjected to EE (73, 88). One study analyzed the effect of EE combined with memantine and found that both treatments enhanced spatial learning and memory in senescence-accelerated mice, and that this effect was stronger when the two treatments were combined than either alone (70, 93), who studied the combined effect of EE and magnesium intake, also reported an amelioration of memory deficits in a transgenic model.
According to the study of Jeon and coworkers, EE compensates the effects of stress on cognitive impairment (91). Peterman et al. (29) concluded that EE does not prevent isolation-induced cognitive deficits in contextual fear-conditioning tests, in contrast to the results of Cao et al. (84), which suggest that the physical component of EE is able to reduce these isolation-induced deficits in cognition. Lastly, exploratory and locomotor behavior have been less studied, with one study reporting that EE enhanced both, while reducing anxiety-like behavior (71). However, the EE paradigm shows inconsistent effects on some anxiety-related responses (72, 82, 108).
To sum up, the environment directly and indirectly impacts on various behavioral processes at multiple layers. Many different mechanistic perspectives on this paradigm are possible and no single factor appears to explain the complexity of the EE model, pointing to the need to consider a more integrative approach to the study of the mediators that give rise to the reported effects (44). Cognitive changes occur mainly via the alteration of neuroplasticity, i.e., neural factors that support behavioral changes. These allow for improvements in functions such as learning and cognition, a desirable outcome for elderly people at risk or already suffering from a neurodegenerative disease (80), or even to promote successful aging in the absence of the disease (32).
EE on synaptic architecture and functional plasticity
The strategies that an individual chooses can determine the construction of cognitive reserve along life. This points to the modifiable nature of healthy factors and strategies that intervene in preventing cognitive decline and aid neuroprotection (32, 53, 92). Experience has the capacity to modify brain structures, building the shelter needed for maintaining brain function and increasing brain resilience to future pathological and/or natural brain damage (1, 17). Undoubtedly one of the key underlying mechanisms responsible for the long-lasting structural and functional changes that the brain undergoes upon exposure to an enriched environment is neuroplasticity, a concept derived from Hebb’s rule (“neurons that fire together, wire together” (45)). This important concept pieced together the functional networking capacity of neurons with a cognitive process, associative learning. It is the synapses that ultimately sense the firing together of the neurons and connect them. In doing so, they undergo structural plasticity, which involves morphological changes in synapses and dendritic spines, and functional plasticity, which emphasizes the physiological changes that accompany the structural plasticity (109). These modifications can be short- or long-termed, and manifested as diminished or enhanced activity of the synapse (short- and long-term depression (STD or LTD) or potentiation (STP or LTP), respectively) (110). The correlation is quite dramatic: LTP promotes the formation of multiple spines between a single axon and a single dendrite (111) and leads to the enlargement of the dendritic spine (112), concomitant with increases in its F-actin content (113) and changes in the actin free monomer/polymeric actin ratio (114) that modify the shape of the spine, which acquires a mushroom-like appearance (115). LTD, in contrast, is associated with the reversible shrinkage (116) and thining of the dendritic spine (115, 117). The most important aspect of synaptic plasticity is that it is required for spatial memory and learning (118).
In animal studies following EE exposure, several outcomes have been reported over the years: increased dendritic branching and length (119), augmented size of dendritic spines and synapses (120), dentate gyrus neurogenesis (121), LTP (122), induced expression of growth factors (123, 124), incresed synaptic protein biosynthesis (125), including specific neurotransmitter receptor subunits (126), resulting in improvements in learning and memory at the behavioral level, even under neuropathological conditions as found in AD (127).
In humans, studies are carried out mainly using functional approaches that report on the connectivity of brain regions, e.g., functional magnetic resonance imaging (fMRI) studies. Increased activity in certain areas of the brain, like the left frontal cortex, is associated with preserved cognitive functions such as memory, and higher level of protective factors, such as education or IQ, in advanced age and AD (128). Neural modifications are more robust if a stimulating environment is applied during specific time windows, i.e., critical periods in postnatal life during which the brain is hyperplastic and sensitive to acquire adaptive signals from external cues (129). However, even during the less sensitive span of adulthood, the brain is still capable of external-induced plastic changes (2). Studies from EE suggest that even in the presence of severe AD neuropathology, environmental stimulation could be a feasible complement to current therapeutic approaches, since even at advanced stages of disease, an enriched environment can still improve cognitive function (130). Animal studies have prompted efforts to apply the constructive principles of the EE paradigm in AD patients, to design appropriate rehabilitation schemes to improve their quality of life (99).
Among the main findings of EE studies, it has been reported that EE prevents cognitive impairment in a transgenic AD model by a mechanism that enhances secretion of exosomal microRNA from the choroid plexus, inhibits astrocytic inflammation, and increases the number of synapses and their density in the subiculum (37). Wild-type mice injected with Aβ oligomers and subsequently subjected to prolonged EE showed increased branching complexity of their neurons in the dentate gyrus (36). Some authors have observed changes in synaptic markers induced by EE in healthy aging mice, but not in transgenic mice: the latter may have been studied at stages when irreversible AD-related pathology had already occurred (88). Barak et al. (78) found an EE-induced improvement of synaptic function as reflected in an increase in the level of the presynaptic protein synaptophysin, an essential molecule for normal synaptic neurotransmission, and a decrease in tomosyn, a protein that inhibits synaptic neurotransmission. Cao et al. (84) also showed an increase in synaptic proteins in mice reared under EE conditions. EE has also been reported to produce an increase in synaptic density in the CA1 area of the hippocampus, suggesting that this area may be capable of experience-dependent plasticity in response to EE despite the presence of accumulating Aβ (88). One study reported that the regulation of synaptic plasticity in the hippocampus was accompanied by a reduction in the expression of APP and a diminution of the deposition of Aβ, thus mitigating the behavioral manifestations of cognitive decline and delaying the onset of AD (64). In a recent study, EE was found not to affect the dendritic architecture or spine density of newborn neurons in wild-type or transgenic mice but promote dendritic development and appearance of of newborn neurons in the dentate gyrus of transgenic mice 6 months after undergoing an EE regime (16). This series of studies strongly suggests that EE impinges on synapse number and distribution, and that changes in synaptic plasticity may be one of the mechanisms underlying the beneficial effects of EE on AD, a subject that calls for further basic neurobiological research.
Given the plastic properties of synapses, they are key candidates as targets of environmental stimulation at the ultrastructural level. Furthermore, synapse loss is a strong correlate of cognitive decline in AD (46, 131, 132) (Figure 3). This section summarized the main conclusions of studies describing changes in synaptic plasticity upon EE in animal models of AD. Current therapeutic approaches aim at restoring neuroplasticity via environmental interventions that may help delay the onset of cognitive decline and dementia (131, 133–135).

In a mouse model of AD, mice exposed to standard housing show lower levels of dendritic arborization, paucity of dendritic spines and less efficient synaptic transmission
In contrast, EE promotes changes in brain cytoarchitecture and synaptic plasticity, as it increases dendritic arborization, dendritic spine size and density, and enhances synaptic plasticity and transmission.
Cognitive reserve associated with environmental enrichment: from rodent experiments to human behavior
It has been widely proposed that a healthy lifestyle and environmental factors have an important influence on cognitive health, triggering interest in the biological mechanisms underlying cognitive, social, and physical activities in the human brain (10). Despite its importance, the impact of these factors on brain neurobiology has been poorly investigated to date, due to obvious limitations on human experimentation (136). Here we propose cognitive reserve as the mechanism whereby experimental outcomes in animals can be extrapolated to human behavior.
In human behavior, the term cognitive reserve, initially used in the context of AD, is often applied in reference to the lack of correlation between the degree of cerebral lesion or pathology and the manifestations of clinical symptoms (17, 137) owing to the use of compensatory cognitive abilities (15, 19). In normal aging, the cognitive reserve accumulated by individuals from childhood to adulthood -even at an advanced age- delays the manifestation of decline and supports normal cognitive function. For those individuals already presenting biomarkers of diseases such as AD, cognitive reserve helps to delay the onset of symptoms. Cognitive reserve is also a functional ability, and though mediated by biological factors, is largely attributable to the activities carried out by the individual. In other words, cognitive reserve strongly depends on the context and life experiences of the individual (11, 17, 138).
The term cognitive reserve is subject to different interpretations. Cabeza and coworkers (139, 140) define reserve as a broad dynamic mechanism through which the brain can accumulate neural resources with the potential to mitigate the effects of neural decline caused by aging or age-related diseases. This mechanism is one of the processes purported to mediate some of the effects of the interaction between genetic background and environmental influence on aging, at both the structural and functional levels. Cabeza and coworkers disagree with the definition proposed by Stern and coworkers, who divide the concept of reserve into cognitive reserve and brain reserve (17, 44).
Cognitive reserve accounts for the functional processes (i.e., networks of brain regions and patterns of interactions between them) that underlie the differential susceptibility of cognitive abilities to brain aging and pathology, whereas brain reserve is put on a qualitatively different level, referring to the neurobiological capital (i.e., variations in structural factors such as synaptic density) that allows some people to better cope with aging and brain pathology (11, 17, 139, 140). Stern and coworkers suggest that cognitive reserve remains a black box, and animal models will be required to elucidate it at the neurobiological level. Animal studies are able to directly address the main questions about cognitive and brain reserve, making experimental approaches a way to link the two concepts and unify the term “reserve”.
Despite the above lack of consensus on the operational definitions of reserve(s), there is general agreement on the role of engaging in stimulating activities such as a higher educational and/or occupational attainment and cognitive leisure activities to promote structural and functional modification of the human brain, leading to the buildup of cognitive reserve that can diminish the manifestations of cognitive decline and dementia. Thus a person with biomarkers for a disease like AD but who has a high cognitive reserve can delay the onset of symptoms or reduce the rate of decline, as compared to a person with the same level of neuropathology but with a lower cognitive reserve (141). For this reason, reserve is proposed as an essential concept for understanding the link between the experimental/biological measures and human behavior. For the purpose of the current review the two concepts are taken as synonymous, as it is our understanding that all cognition depends on the healthy function and structure of the brain, and the two aspects cannot be separated. However, our preference is to use “cognitive reserve”, as it emphasizes the behavioral/functional aspect of the reserve.
The concept “reserve-builders” refers to the activities that enhance cognitive reserve. The more intensely and assiduously such activities are pursued along an indiviudual’s lifespan, the higher are the chances of building up cognitive reserve and the lower the risk of developing AD or other forms of dementia. Reserve builders are considered proxy measures of the components that are stimulated in EE. Reserve-builders that stimulate the cognitive component are activities such as reading, painting, poetry, song writing, sculpture, and attending lectures and discussions. Premorbid IQ and years and type of schooling and occupational attainment are considered static proxy measures of the cognitive component. Reserve-builders that promote physical stimulation in humans are activities such as running, trekking, biking, swimming, dancing, doing yoga and skiing (95). Physical exercise in general is beneficial for the functional autonomy of patients with cognitive impairment and/or dementia, with significant benefits for cognition being attributed to aerobic exercise in particular. Aerobic exercise promotes adherence and compliance and is largely preferred by patients over balance, flexibility or strength exercises. Patients with dementia or those at risk of developing dementia should therefore be encouraged to undertake aerobic exercise, with a focus on the patient’s adherence rather than on a fixed amount of exercise (142). Despite WHO recommendations to engage in a daily routine of physical activity and exercise to prevent sedentarism and avoid the development of risk factors such as diabetes and cardiovascular diseases, many elderly people particularly in developing countries do not have an active lifestyle. The above-mentioned risk factors in turn increase the chances of developing AD, which is why one of the therapeutic goals is to encourage older adults to gradually incorporate physical exercise in their daily routine (143). Reserve-builders of the social component are activities like playing structured games, volunteering, and social activities (95). Clinical trials assessing the effect of social interventions have concluded that being socially active correlates with a higher prevention of cognitive decline, whereas social isolation is correlated with a greater risk of developing AD. As a powerful protective factor, stimulation of the social component must be considered for patients as well as for people in care homes (144, 145). Other factors promoting cognitive reserve are for example bilingualism, continuing education, a healthy lifestyle including the type of diet, sleep hygiene, and limiting caffeine or nicotine (95). In the specific case of AD, cognitive reserve is a key variable that links AD neuropathology with loss of cognitive function (10). Stimulating activities are associated with a lesser risk of developing dementia (4, 32); however, risk factors such as psychosocial stressors, inappropriate medication, or other comorbid pathologies may reduce cognitive reserve capacity and accelerate the onset of symptomatic AD (20). One study suggested that improvement in vascular function, especially in hypertensive individuals, may account for the delayed cognitive decline in patients with mild cognitive impairment, after participating in a 7-month training program aimed at stimulating cognitive, social and physical factors (146). Another longitudinal study associated a cognitively active lifestyle with a delay in the onset of AD dementia by 5 years, whereas less cognitively active individuals presented an earlier age of onset (147). This means that the decline in cognitive performance is a convolution of multiple protective factors combined with risk factors that either ameliorate or harm cognitive and brain health. From a biological standpoint, cognitive reserve may reflect increased neuronal connectivity in brain regions involved in cognitive processes like learning and memory, and which respond to the healthy lifestyle and enriched environment, compensating and withstanding detrimental pathophysiological mechanisms of dementia before they manifest clinically (17).
Interventions addressing lifestyle improvements in general, i.e. covering multiple risk and protective factors, have a higher probability of preventing cognitive impairment effectively. For example, the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER study) is a multicomponent intervention that considers multiple lifestyle factors and serves as a guide for combination therapy trials, which integrate non-pharmacological, pharmacological, and nutrition-based approaches. The underlying hypothesis is that as AD and dementia are of a multifactorial nature, simultaneous targets of intervention are needed to promote cognitive reserve and prevent undesired symptoms, achieved by addressing lifestyle factors such as diet, exercise, cognitive training, and vascular risk monitoring (148–150). It has been shown that this intervention is effective regardless of individual differences in cardiovascular conditions, baseline cognition, sociodemographic or socioeconomic status (150). Based on the FINGER study, other multidomain lifestyle interventions have been designed and some have also been adapted for application in virtual (digital) contexts, all of them showing significant potential to enhance cognitive reserve (151). Other programs aimed at enhancing cognitive performance in patients already suffering cognitive impairment have also shown positive results. The combination of cognitive and physical stimulation appears to be most effective towards this end (152–154). However, non-pharmacological treatments can be combined with pharmacological treatments as a broader therapeutic strategy. Acetylcholinesterase inhibitors are the most frequently used drugs to diminish cognitive symptoms in AD. Hence, drug therapy is usually combined with stimulation therapy, which most commonly includes physical and/or cognitive stimulation. The combined stimulation + drug treatment has a significant positive impact on cognitive performance in contrast to pharmacological treatment alone, offering a promising therapeutic strategy to enhance cognitive reserve and delay the onset and progression of AD (155, 156). Additionally, these strategies could be combined with novel emerging drugs that are aimed not at curing the disease but at improving its course indirectly (like statins in the primary prevention of vascular risk factors (157)), and nutritional supplements such as Ginkgo biloba (158) or vitamin E (159); however, these approaches are not currently recommended due to their low proven efficiency (160).
Environmental stimuli can also have positive effects on those already suffering dementia: for example, one study demonstrated that enriched gardens (which stimulate cognition, independence in daily activities and motor functions) can improve cognitive and motor abilities, in contrast to standard (i.e., not stimulating) gardens (161). Other authors carried out longitudinal studies on the effects of cognitive reserve on clinical AD progression, finding that a higher cognitive reserve, assessed through a quantitative scale, was associated with lower rates of conversion to dementia, thus concluding that reserve can affect the progression of the disease by delaying its onset. Patients with higher levels of cognitive reserve showed a more pronounced decline once the cognitive deterioration began, whereas those with lesser cognitive reserve had an earlier but more gradual decrease in cognitive functioning (162). Thus the neurobiological mechanism by which cognitive reserve ameliorates the manifestation of cognitive decline and dementia appears to be exerted through the development of stronger neuronal connections that can withstand a greater amount of neuropathology before manifesting the behavioral symptoms of the disease (128). Neurobiological studies in human tissue are a needy area, particularly with regard to the protective role of cognitive reserve against cognitive decline and dementia (163).
Concluding remarks
The incidence of AD is increasing at an alarming pace, leading to concerns about the ensuing social, biomedical and financial burdens. The urge to contain or ameliorate the tempo of this neurodegenerative disease is clear, but our ignorance about its etiopathology has precluded efficient interventions. The combination of lifestyle and environmental adjustments offers promising strategies.
Special focus should be given to therapeutic approaches that help restore or enhance neuroplasticity, which is altered in AD, and to studying the main mechanisms underlying the benefits that environmental interventions bring to cognitive health. The complexity of biological interactions with environmental factors is far from being understood, and the basis of processes that contribute towards delays in cognitive decline and dementia need to be studied in greater depth. Comprehending the multiple biological bases underlying human cognitive reserve and its deterioration in AD, in particular relating to neuroplasticity, and the way in which these properties can be modified by environmental factors, is an important step towards developing more efficient prophylactic and therapeutic strategies in AD and other forms of dementia.
References
Puderbaugh, M. and P.D. Emmady, Neuroplasticity, in StatPearls. 2022, StatPearls Publishing, Copyright © 2022, StatPearls Publishing LLC.: Treasure Island (FL).
Kühn, S. and U. Lindenberger, Chapter 6. Research on Human Plasticity in Adulthood. 2016.
Chen, X., J. Hu, and A. Sun, The Beneficial Effect of Enriched Environment on Pathogenesis of Alzheimer’s Disease. Yangtze Medicine, 2018. 02: p. 225–243.
Phillips, C., Lifestyle Modulators of Neuroplasticity: How Physical Activity, Mental Engagement, and Diet Promote Cognitive Health during Aging. Neural Plast, 2017. 2017: p. 3589271.
Janssen, H., et al., Translating the use of an enriched environment poststroke from bench to bedside: study design and protocol used to test the feasibility of environmental enrichment on stroke patients in rehabilitation. Int J Stroke, 2012. 7(6): p. 521–6.
Organization, W.H. Dementia. 2021; Available from: https://www.who.int/news-room/fact-sheets/detail/dementia#:~:text=Worldwide%2C%20around%2055%20million%20people,and%20139%20million%20in%202050.
Bekdash, R.A., The Cholinergic System, the Adrenergic System and the Neuropathology of Alzheimer’s Disease. Int J Mol Sci, 2021. 22(3).
Tatulian, S.A., Challenges and hopes for Alzheimer’s disease. Drug Discov Today, 2022. 27(4): p. 1027–1043.
Perneczky, R., et al., Translational research on reserve against neurodegenerative disease: consensus report of the International Conference on Cognitive Reserve in the Dementias and the Alzheimer’s Association Reserve, Resilience and Protective Factors Professional Interest Area working groups. BMC Medicine, 2019. 17(1): p. 47.
Arenaza-Urquijo, E.M., M. Wirth, and G. Chételat, Cognitive reserve and lifestyle: moving towards preclinical Alzheimer’s disease. Front Aging Neurosci, 2015. 7: p. 134.
Stern, Y., et al., Mechanisms underlying resilience in ageing. Nature Reviews Neuroscience, 2019. 20(4): p. 246–246.
Mercerón-Martínez, D., et al., Alzheimer’s Disease, Neural Plasticity, and Functional Recovery. J Alzheimers Dis, 2021. 82(s1): p. S37–S50.
Harrison, S.L., et al., Exploring strategies to operationalize cognitive reserve: A systematic review of reviews. Journal of Clinical and Experimental Neuropsychology, 2015. 37(3): p. 253–264.
Cancino, M. and L. Rehbein, Factores de riesgo y precursores del Deterioro Cognitivo Leve (DCL): Una mirada sinóptica % J Terapia psicológica. 2016. 34: p. 183–189.
Sampedro-Piquero, P. and A. Begega, Environmental enrichment as a positive behavioral intervention across the lifespan. Current Neuropharmacology, 2017. 15(4): p. 459–470.
Zhang, Y., et al., The short-term improvements of enriched environment in behaviors and pathological changes of APP/PS1 mice via regulating cytokines. Hum Vaccin Immunother, 2018. 14(8): p. 2003–2011.
Stern, Y., et al., Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimers Dement, 2020. 16(9): p. 1305–1311.
Torres-Lista, V. and L. Giménez-Llort, Early postnatal handling and environmental enrichment improve the behavioral responses of 17-month-old 3xTg-AD and non-transgenic mice in the Forced Swim Test in a gender-dependent manner. Behavioural Processes, 2015. 120: p. 120–127.
Shepherd, A., et al., Transgenic Mouse Models as Tools for Understanding How Increased Cognitive and Physical Stimulation Can Improve Cognition in Alzheimer’s Disease. Brain Plasticity, 2018. 4: p. 127–150.
Moga, D.C., et al., INtervention for Cognitive Reserve Enhancement in delaying the onset of Alzheimer’s Symptomatic Expression (INCREASE), a randomized controlled trial: rationale, study design, and protocol. Trials, 2019. 20(1): p. 806.
Lei, P., S. Ayton, and A.I. Bush, The essential elements of Alzheimer’s disease. J Biol Chem, 2021. 296: p. 100105.
Mahaman, Y.A.R., et al., Biomarkers used in Alzheimer’s disease diagnosis, treatment, and prevention. Ageing Res Rev, 2022. 74: p. 101544.
Budelier, M.M. and R.J. Bateman, Biomarkers of Alzheimer Disease. J Appl Lab Med, 2020. 5(1): p. 194–208.
Zangrossi, A., et al., Heterogeneity and Factorial Structure in Alzheimer’s Disease: A Cognitive Perspective. J Alzheimers Dis, 2021. 83(3): p. 1341–1351.
Cloak, N. and Y. Al Khalili, Behavioral And Psychological Symptoms In Dementia, in StatPearls. 2022, StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.: Treasure Island (FL).
Boublay, N., A.M. Schott, and P. Krolak-Salmon, Neuroimaging correlates of neuropsychiatric symptoms in Alzheimer’s disease: a review of 20 years of research. Eur J Neurol, 2016. 23(10): p. 1500–9.
Ismail, Z., et al., Psychosis in Alzheimer disease — mechanisms, genetics and therapeutic opportunities. Nat Rev Neurol, 2022. 18(3): p. 131–144.
Rodríguez-Blázquez, C., et al., Calidad de vida y estado de salud en personas mayores de 60 años con demencia institucionalizadas % J Revista Española de Salud Pública. 2015. 89: p. 51–60.
Peterman, J.L., et al., Prolonged isolation stress accelerates the onset of Alzheimer’s disease-related pathology in 5xFAD mice despite running wheels and environmental enrichment. Behav Brain Res, 2020. 379: p. 112366.
Calabrò, M., et al., The biological pathways of Alzheimer disease: a review. AIMS Neurosci, 2021. 8(1): p. 86–132.
Montarolo, F., et al., Early enriched environment exposure protects spatial memory and accelerates amyloid plaque formation in APP(Swe)/PS1(L166P) mice. PLoS One, 2013. 8(7): p. e69381.
Mora, F., Successful brain aging: plasticity, environmental enrichment, and lifestyle. Dialogues Clin Neurosci, 2013. 15(1): p. 45–52.
Ziegler-Waldkirch, S., et al., Environmental enrichment reverses Aβ pathology during pregnancy in a mouse model of Alzheimer’s disease. Acta Neuropathol Commun, 2018. 6(1): p. 44.
Kuo, Y.-C. and R. Rajesh, Challenges in the treatment of Alzheimer’s disease: recent progress and treatment strategies of pharmaceuticals targeting notable pathological factors. Expert Review of Neurotherapeutics, 2019. 19(7): p. 623–652.
Cappa, S.F., The Quest for an Alzheimer Therapy. Front Neurol, 2018. 9: p. 108.
Xu, H., et al., Environmental Enrichment Potently Prevents Microglia-Mediated Neuroinflammation by Human Amyloid β-Protein Oligomers. J Neurosci, 2016. 36(35): p. 9041–56.
Nakano, M., An enriched environment improves cognitive impairment in an Alzheimer’s disease model by enhancing the secretion of exosomal miR-146a from the choroid plexus. 2020. 16(S2): p. e041682.
Mandolesi, L., et al., Environmental Factors Promoting Neural Plasticity: Insights from Animal and Human Studies. Neural Plast, 2017. 2017: p. 7219461.
Prado Lima, M.G., et al., Environmental enrichment and exercise are better than social enrichment to reduce memory deficits in amyloid beta neurotoxicity. Proc Natl Acad Sci U S A, 2018. 115(10): p. E2403–E2409.
Hüttenrauch, M., et al., Physical activity delays hippocampal neurodegeneration and rescues memory deficits in an Alzheimer disease mouse model. Transl Psychiatry, 2016. 6(5): p. e800.
Bisht, K., K. Sharma, and M. Tremblay, Chronic stress as a risk factor for Alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiol Stress, 2018. 9: p. 9–21.
Snowdon, D.A., Healthy aging and dementia: findings from the Nun Study. Ann Intern Med, 2003. 139 (5 Pt 2): p. 450–4.
Bauckneht, M., et al., Metabolic correlates of reserve and resilience in MCI due to Alzheimer’s Disease (AD). Alzheimers Res Ther, 2018. 10(1): p. 35.
Kempermann, G., Environmental enrichment, new neurons and the neurobiology of individuality. Nat Rev Neurosci, 2019. 20(4): p. 235–245.
Hebb, D.O., The organization of behavior; a neuropsychological theory. The organization of behavior; a neuropsychological theory. 1949, Oxford, England: Wiley. xix, 335-xix, 335.
Nithianantharajah, J. and A.J. Hannan, Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci, 2006. 7(9): p. 697–709.
van Dellen, A., et al., Delaying the onset of Huntington’s in mice. Nature, 2000. 404(6779): p. 721–2.
Hockly, E., et al., Environmental enrichment slows disease progression in R6/2 Huntington’s disease mice. 2002. 51(2): p. 235–242.
Spires, T.L., et al., Environmental enrichment rescues protein deficits in a mouse model of Huntington’s disease, indicating a possible disease mechanism. J Neurosci, 2004. 24(9): p. 2270–6.
Arendash, G.W., et al., Environmental enrichment improves cognition in aged Alzheimer’s transgenic mice despite stable beta-amyloid deposition. Neuroreport, 2004. 15(11): p. 1751–4.
Robison, L.S., et al., Environmental Enrichment: Disentangling the Influence of Novelty, Social, and Physical Activity on Cerebral Amyloid Angiopathy in a Transgenic Mouse Model. Int J Mol Sci, 2020. 21(3).
Rodriguez, A., et al., Relación entre la Reserva Cognitiva y el Enriquecimiento Ambiental: Una revisión del Aporte de las Neurociencias a la comprensión del Envejecimiento Saludable. Cuadernos de Neuropsicología / Panamerican Journal of Neuropsychology, 2014. 8(2): p. 171–201.
Bezzina, C., et al., Environmental enrichment does not influence hypersynchronous network activity in the Tg2576 mouse model of Alzheimer’s disease. Front Aging Neurosci, 2015. 7: p. 178.
Hu, Y.S., et al., Molecular mechanisms of environmental enrichment: impairments in Akt/GSK3β, neurotrophin-3 and CREB signaling. PLoS One, 2013. 8(5): p. e64460.
Drummond, E. and T. Wisniewski, Alzheimer’s disease: experimental models and reality. Acta Neuropathol, 2017. 133(2): p. 155–175.
Barrett, J.E. and P. Mcgonigle. Rodent Models for Alzheimer’s Disease in Drug Discovery. 2017.
Justice, M.J. and P. Dhillon, Using the mouse to model human disease: increasing validity and reproducibility. Dis Model Mech, 2016. 9(2): p. 101–3.
Elder, G.A., M.A. Gama Sosa, and R. De Gasperi, Transgenic mouse models of Alzheimer’s disease. Mt Sinai J Med, 2010. 77(1): p. 69–81.
Holcomb, L., et al., Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med, 1998. 4(1): p. 97–100.
Oddo, S., et al., Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron, 2003. 39(3): p. 409–21.
Oakley, H., et al., Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. 2006. 26(40): p. 10129–10140.
Radde, R., et al., Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep, 2006. 7(9): p. 940–6.
Jankowsky, J.L., et al., APP processing and amyloid deposition in mice haplo-insufficient for presenilin 1. Neurobiol Aging, 2004. 25(7): p. 885–92.
Li, J.Z., et al., An enriched environment delays the progression from mild cognitive impairment to Alzheimer’s disease in senescence-accelerated mouse prone 8 mice. Exp Ther Med, 2021. 22(5): p. 1320.
Brouillette, J., et al., Neurotoxicity and memory deficits induced by soluble low-molecular-weight amyloid-β1–42 oligomers are revealed in vivo by using a novel animal model. J Neurosci, 2012. 32(23): p. 7852–61.
Lahiani-Cohen, I., et al., Moderate environmental enrichment mitigates tauopathy in a neurofibrillary tangle mouse model. J Neuropathol Exp Neurol, 2011. 70(7): p. 610–21.
Xu, H., et al., Enriched environment enhances β-adrenergic signaling to prevent microglia inflammation by amyloid-β. EMBO Mol Med, 2018. 10(9).
Li, S., et al., Environmental novelty activates β2-adrenergic signaling to prevent the impairment of hippocampal LTP by Aβ oligomers. Neuron, 2013. 77(5): p. 929–41.
Wei, Z., et al., Environmental enrichment prevents Aβ oligomer-induced synaptic dysfunction through mirna-132 and hdac3 signaling pathways. Neurobiol Dis, 2020. 134: p. 104617.
Dong, J., et al., Memantine combined with environmental enrichment improves spatial memory and alleviates Alzheimer’s disease-like pathology in senescence-accelerated prone-8 (SAMP8) mice. J Biomed Res, 2012. 26(6): p. 439–47.
Görtz, N., et al., Effects of environmental enrichment on exploration, anxiety, and memory in female TgCRND8 Alzheimer mice. Behav Brain Res, 2008. 191(1): p. 43–8.
Pietropaolo, S., J. Feldon, and B.K. Yee, Environmental enrichment eliminates the anxiety phenotypes in a triple transgenic mouse model of Alzheimer’s disease. Cognitive, Affective & Behavioral Neuroscience, 2014. 14(3): p. 996–1008.
Stuart, K.E., et al., Late-life environmental enrichment preserves short-term memory and may attenuate microglia in male APP/PS1 mice. Neuroscience, 2019. 408: p. 282–292.
Stuart, K.E., et al., Mid-life environmental enrichment increases synaptic density in CA1 in a mouse model of Aβ-associated pathology and positively influences synaptic and cognitive health in healthy ageing. J Comp Neurol, 2017. 525(8): p. 1797–1810.
Jankowsky, J.L., et al., Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Neuropathol Exp Neurol, 2003. 62(12): p. 1220–7.
Herring, A., et al., Reduction of cerebral oxidative stress following environmental enrichment in mice with Alzheimer-like pathology. Brain Pathol, 2010. 20(1): p. 166–75.
Herring, A., et al., Environmental Enrichment Counteracts Alzheimer’s Neurovascular Dysfunction in TgCRND8 Mice. 2008. 18(1): p. 32–39.
Barak, B., et al., Opposing actions of environmental enrichment and Alzheimer’s disease on the expression of hippocampal microRNAs in mouse models. Transl Psychiatry, 2013. 3(9): p. e304.
Cotel, M.C., et al., Environmental enrichment fails to rescue working memory deficits, neuron loss, and neurogenesis in APP/PS1KI mice. Neurobiol Aging, 2012. 33(1): p. 96–107.
Griñán-Ferré, C., et al., Environmental Enrichment Improves Cognitive Deficits, AD Hallmarks and Epigenetic Alterations Presented in 5xFAD Mouse Model. Front Cell Neurosci, 2018. 12: p. 224.
Polito, L., et al., Environmental enrichment lessens cognitive decline in APP23 mice without affecting brain sirtuin expression. J Alzheimers Dis, 2014. 42(3): p. 851–64.
Stazi, M. and O. Wirths, Physical activity and cognitive stimulation ameliorate learning and motor deficits in a transgenic mouse model of Alzheimer’s disease. Behav Brain Res, 2021. 397: p. 112951.
Berardi, N., et al., Environmental enrichment delays the onset of memory deficits and reduces neuropathological hallmarks in a mouse model of Alzheimer-like neurodegeneration. J Alzheimers Dis, 2007. 11(3): p. 359–70.
Cao, W.Y., et al., Role of early environmental enrichment on the social dominance tube test at adulthood in the rat. Psychopharmacology (Berl), 2017. 234(22): p. 3321–3334.
Herring, A., et al., Preventive and therapeutic types of environmental enrichment counteract beta amyloid pathology by different molecular mechanisms. Neurobiol Dis, 2011. 42(3): p. 530–8.
Herring, A., et al., Environmental enrichment enhances cellular plasticity in transgenic mice with Alzheimer-like pathology. Exp Neurol, 2009. 216(1): p. 184–92.
Rodríguez, J.J., H.N. Noristani, and A. Verkhratsky, Microglial response to Alzheimer’s disease is differentially modulated by voluntary wheel running and enriched environments. Brain Struct Funct, 2015. 220(2): p. 941–53.
Stuart, K.E., et al., Mid-Life Complex and Novel Environmental Enrichment Increase Corticosterone and Exacerbate AB Neuropathology in a Mouse Model of Alzheimer’s Disease. 2016. 12 (7, Supplement 1): p. P1039–P1040.
Arranz, L., et al., Effect of environmental enrichment on the immunoendocrine aging of male and female triple-transgenic 3xTg-AD mice for Alzheimer’s disease. J Alzheimers Dis, 2011. 25(4): p. 727–37.
Arranz, L., et al., Environmental enrichment improves age-related immune system impairment: long-term exposure since adulthood increases life span in mice. Rejuvenation research, 2010. 13(4): p. 415–428.
Jeong, Y.H., et al., Environmental enrichment compensates for the effects of stress on disease progression in Tg2576 mice, an Alzheimer’s disease model. J Neurochem, 2011. 119(6): p. 1282–93.
Balthazar, J., et al., Enriched Environment Significantly Reduced Senile Plaques in a Transgenic Mice Model of Alzheimer’s Disease, Improving Memory. Front Aging Neurosci, 2018. 10: p. 288.
Huang, Y., et al., Magnesium boosts the memory restorative effect of environmental enrichment in Alzheimer’s disease mice. 2018. 24(1): p. 70–79.
Kalogeraki, E., J. Pielecka-Fortuna, and S. Löwel, Environmental enrichment accelerates ocular dominance plasticity in mouse visual cortex whereas transfer to standard cages resulted in a rapid loss of increased plasticity. PLoS One, 2017. 12(10): p. e0186999.
Serra, L., et al., Rethinking the Reserve with a Translational Approach: Novel Ideas on the Construct and the Interventions. J Alzheimers Dis, 2018. 65(4): p. 1065–1078.
Stozicka, Z., et al., Environmental Enrichment Rescues Functional Deficit and Alters Neuroinflammation in a Transgenic Model of Tauopathy. Journal of Alzheimer’s disease: JAD, 2020. 74(3): p. 951–964.
Mirochnic, S., et al., Age effects on the regulation of adult hippocampal neurogenesis by physical activity and environmental enrichment in the APP23 mouse model of Alzheimer disease. Hippocampus, 2009. 19(10): p. 1008–18.
Rodríguez, J.J., et al., Voluntary running and environmental enrichment restores impaired hippocampal neurogenesis in a triple transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res, 2011. 8(7): p. 707–17.
Verret, L., et al., Transient enriched housing before amyloidosis onset sustains cognitive improvement in Tg2576 mice. Neurobiol Aging, 2013. 34(1): p. 211–25.
Jankowsky, J.L., et al., Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci, 2005. 25(21): p. 5217–24.
Maesako, M., et al., Environmental enrichment ameliorated high-fat diet-induced Aβ deposition and memory deficit in APP transgenic mice. Neurobiol Aging, 2012. 33(5): p. 1011.e11–23.
Maurer, S.V. and C.L. Williams, The Cholinergic System Modulates Memory and Hippocampal Plasticity via Its Interactions with Non-Neuronal Cells. Front Immunol, 2017. 8: p. 1489.
Hampel, H., et al., Reply: Optimal use of cholinergic drugs in Alzheimer’s disease. Brain, 2018. 141(9): p. e69–e69.
Vallés, A.S. and F.J. Barrantes, Dendritic spine membrane proteome and its alterations in autistic spectrum disorder. Adv Protein Chem Struct Biol, 2022. 128: p. 435–474.
Barrantes, F.J., V. Borroni, and S.J.F.l. Vallés, Neuronal nicotinic acetylcholine receptor-cholesterol crosstalk in Alzheimer’s disease. 2010. 584(9): p. 1856–1863.
Valles, A.S., Targeting Brain alpha;7 Nicotinic Acetylcholine Receptors in Alzheimer’s Disease: Rationale and Current Status, ed. M.V. Borroni and F.J. Barrantes. 2014.
Beauquis, J., et al., Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease. Exp Neurol, 2013. 239: p. 28–37.
Blázquez, G., et al., Cognitive and emotional profiles of aged Alzheimer’s disease (3xTgAD) mice: effects of environmental enrichment and sexual dimorphism. Behav Brain Res, 2014. 268: p. 185–201.
Yuste, R. and T. Bonhoeffer, Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu.Rev.Neurosci., 2001. 24: p. 1071–1089.
Bear, M.F. and R.C. Malenka, Synaptic plasticity: LTP and LTD. Curr.Opin. Neurobiol., 1994. 4(3): p. 389–399.
Toni, N., et al., LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature, 1999. 402(6760): p. 421–425.
Lang, C., et al., Transient expansion of synaptically connected dendritic spines upon induction of hippocampal long-term potentiation. Proc.Natl.Acad. Sci.U.S.A, 2004. 101(47): p. 16665–16670.
Fukazawa, Y., et al., Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron, 2003. 38(3): p. 447–460.
Bonilla-Quintana, M., et al., Actin in Dendritic Spines Self-Organizes into a Critical State. 2020: p. 2020.04.22.054577.
Medvedev, N.I., et al., The N-methyl-D-aspartate receptor antagonist CPP alters synapse and spine structure and impairs long-term potentiation and long-term depression induced morphological plasticity in dentate gyrus of the awake rat. Neuroscience, 2010. 165(4): p. 1170–1181.
Zhou, Q., K.J. Homma, and M.M. Poo, Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron, 2004. 44(5): p. 749–757.
Bi, G.Q. and M.M. Poo, Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci., 1998. 18(24): p. 10464–10472.
Tsien, J.Z., P.T. Huerta, and S. Tonegawa, The Essential Role of Hippocampal CA1 NMDA Receptor’s Dependent Synaptic Plasticity in Spatial Memory. Cell, 1996. 87(7): p. 1327–1338.
Faherty, C.J., D. Kerley, and R.J. Smeyne, A Golgi-Cox morphological analysis of neuronal changes induced by environmental enrichment. Brain Res Dev Brain Res, 2003. 141(1–2): p. 55–61.
Leggio, M.G., et al., Environmental enrichment promotes improved spatial abilities and enhanced dendritic growth in the rat. Behav Brain Res, 2005. 163(1): p. 78–90.
Kempermann, G., H.G. Kuhn, and F.H. Gage, More hippocampal neurons in adult mice living in an enriched environment. Nature, 1997. 386(6624): p. 493–5.
Artola, A., et al., Long-lasting modulation of the induction of LTD and LTP in rat hippocampal CA1 by behavioural stress and environmental enrichment. Eur J Neurosci, 2006. 23(1): p. 261–72.
Pham, T.M., et al., Effects of environmental enrichment on cognitive function and hippocampal NGF in the non-handled rats. Behav Brain Res, 1999. 103(1): p. 63–70.
Ickes, B.R., et al., Long-term environmental enrichment leads to regional increases in neurotrophin levels in rat brain. Exp Neurol, 2000. 164(1): p. 45–52.
Nithianantharajah, J., et al., Environmental enrichment results in cortical and subcortical changes in levels of synaptophysin and PSD-95 proteins. 2004. 81(3): p. 200–210.
Naka, F., et al., Modification of AMPA receptor properties following environmental enrichment. Brain Dev, 2005. 27(4): p. 275–8.
Tang, Y.P., et al., Differential effects of enrichment on learning and memory function in NR2B transgenic mice. Neuropharmacology, 2001. 41(6): p. 779–90.
Franzmeier, N., et al., The left frontal cortex supports reserve in aging by enhancing functional network efficiency. Alzheimer’s Research & Therapy, 2018. 10(1): p. 28.
Moody, L., H. Chen, and Y.X. Pan, Early-Life Nutritional Programming of Cognition-The Fundamental Role of Epigenetic Mechanisms in Mediating the Relation between Early-Life Environment and Learning and Memory Process. Adv Nutr, 2017. 8(2): p. 337–350.
Fischer, A., Environmental enrichment as a method to improve cognitive function. What can we learn from animal models? NeuroImage, 2016. 131: p. 42–47.
Gajewski, P.D., et al., Impact of Biological and Lifestyle Factors on Cognitive Aging and Work Ability in the Dortmund Vital Study: Protocol of an Interdisciplinary, Cross-sectional, and Longitudinal Study. JMIR Res Protoc, 2022. 11(3): p. e32352.
Koch, G. and D. Spampinato, Alzheimer disease and neuroplasticity. Handb Clin Neurol, 2022. 184: p. 473–479.
Foster, P.P., K.P. Rosenblatt, and R.O. Kuljis, Exercise-induced cognitive plasticity, implications for mild cognitive impairment and Alzheimer’s disease. Front Neurol, 2011. 2: p. 28.
De la Rosa, A., et al., Physical exercise in the prevention and treatment of Alzheimer’s disease. J Sport Health Sci, 2020. 9(5): p. 394–404.
Kępka, A., et al., Healthy Food Pyramid as Well as Physical and Mental Activity in the Prevention of Alzheimer’s Disease. 2022. 14(8): p. 1534.
Bartrés-Faz, D., et al., Meaning in life: resilience beyond reserve. Alzheimer’s Research & Therapy, 2018. 10(1): p. 47.
Nilsson, J. and M. Lovdén, Naming is not explaining: future directions for the “cognitive reserve” and “brain maintenance” theories. Alzheimer’s research & therapy, 2018. 10: p. 34.
Stern, Y., Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol, 2012. 11(11): p. 1006–12.
Cabeza, R., et al., Reply to’ Mechanisms underlying resilience in ageing’. Nature Reviews Neuroscience, 2019. 20(4): p. 247–247.
Cabeza, R., et al., Maintenance, reserve and compensation: the cognitive neuroscience of healthy ageing. Nature Reviews Neuroscience, 2018. 19(11): p. 701–710.
Soldan, A., et al., Cognitive reserve and long-term change in cognition in aging and preclinical Alzheimer’s disease. Neurobiol Aging, 2017. 60: p. 164–172.
Fang, Y., Guiding research and practice: a conceptual model for aerobic exercise training in Alzheimer’s disease. Am J Alzheimers Dis Other Demen, 2011. 26(3): p. 184–94.
Amadasi, E., S.S. Rodríguez Espínola, and C.S. Garofalo, Condiciones de vida de las personas mayores (2017–2021): vulnerabilidades en clave de pandemia por COVID-19, EDUCA, Editor. 2022, EDSA Serie Agenda para la Equidad: Repositorio Institucional UCA.
Dyer, A.H., et al., Social networks in mild-to-moderate Alzheimer disease: longitudinal relationships with dementia severity, cognitive function, and adverse events. Aging Ment Health, 2021. 25(10): p. 1923–1929.
Hsiao, Y.H., C.H. Chang, and P.W. Gean, Impact of social relationships on Alzheimer’s memory impairment: mechanistic studies. J Biomed Sci, 2018. 25(1): p. 3.
Bruno, R.M., et al., Vascular Function Is Improved After an Environmental Enrichment Program: The Train the Brain-Mind the Vessel Study. Hypertension, 2018. 71(6): p. 1218–1225.
Wilson, R.S., et al., Cognitive Activity and Onset Age of Incident Alzheimer Disease Dementia. Neurology, 2021. 97(9): p. e922–e929.
Ngandu, T., et al., A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet, 2015. 385(9984): p. 2255–63.
Kivipelto, M., et al., World-Wide FINGERS Network: A global approach to risk reduction and prevention of dementia. Alzheimers Dement, 2020. 16(7): p. 1078–1094.
Rosenberg, A., et al., Multidomain Interventions to Prevent Cognitive Impairment, Alzheimer’s Disease, and Dementia: From FINGER to World- Wide FINGERS. J Prev Alzheimers Dis, 2020. 7(1): p. 29–36.
Bott, N.T., et al., Face-to-Face and Digital Multidomain Lifestyle Interventions to Enhance Cognitive Reserve and Reduce Risk of Alzheimer’s Disease and Related Dementias: A Review of Completed and Prospective Studies. Nutrients, 2019. 11(9).
Li, L., et al., Multi-component exercise training improves the physical and cognitive function of the elderly with mild cognitive impairment: a six-month randomized controlled trial. Ann Palliat Med, 2021. 10(8): p. 8919–8929.
Ten Brinke, L.F., et al., The Effects of Computerized Cognitive Training With and Without Physical Exercise on Cognitive Function in Older Adults: An 8-Week Randomized Controlled Trial. J Gerontol A Biol Sci Med Sci, 2020. 75(4): p. 755–763.
Yang, H.L., et al., Construction and evaluation of multidomain attention training to improve alertness attention, sustained attention, and visual-spatial attention in older adults with mild cognitive impairment: A randomized controlled trial. Int J Geriatr Psychiatry, 2020. 35(5): p. 537–546.
Andersen, F., et al., The effect of stimulation therapy and donepezil on cognitive function in Alzheimer’s disease. A community based RCT with a two-by-two factorial design. BMC Neurol, 2012. 12: p. 59.
Matsuda, O., et al., Short-term effect of combined drug therapy and cognitive stimulation therapy on the cognitive function of Alzheimer’s disease. Psychogeriatrics, 2010. 10(4): p. 167–72.
Wolozin, B., Statins and therapy of Alzheimer’s disease: questions of efficacy versus trial design. Alzheimers Res Ther, 2012. 4(1): p. 3.
Kandiah, N., et al., Treatment of dementia and mild cognitive impairment with or without cerebrovascular disease: Expert consensus on the use of Ginkgo biloba extract, EGb 761®. CNS neuroscience & therapeutics, 2019. 25(2): p. 288–298.
Farina, N., et al., Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst Rev, 2017. 4(4): p. Cd002854.
O’Brien, J.T., et al., Clinical practice with anti-dementia drugs: A revised (third) consensus statement from the British Association for Psychopharmacology. J Psychopharmacol, 2017. 31(2): p. 147–168.
Bourdon, E. and J. Belmin, Enriched gardens improve cognition and independence of nursing home residents with dementia: a pilot controlled trial. Alzheimers Res Ther, 2021. 13(1): p. 116.
Lee, D.H., et al., Effects of Cognitive Reserve in Alzheimer’s Disease and Cognitively Unimpaired Individuals. Front Aging Neurosci, 2021. 13: p. 784054.
Pettigrew, C. and A. Soldan, Defining Cognitive Reserve and Implications for Cognitive Aging. Curr Neurol Neurosci Rep, 2019. 19(1): p. 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical standards: Standards concerning publication ethics were followed.
Conflict of interest: The authors declare to have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Colavitta, M.F., Grasso, L. & Barrantes, F.J. Environmental Enrichment in Murine Models and Its Translation to Human Factors Improving Conditions in Alzheimer Disease. J Prev Alzheimers Dis 10, 287–300 (2023). https://doi.org/10.14283/jpad.2023.5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.14283/jpad.2023.5