
Abstract
Background
Though our understanding of Alzheimer’s disease (AD) remains elusive, it is well known that the disease starts long before the first signs of dementia. This is supported by the large number of symptomatic drug failures in clinical trials and the increased trend to enroll patients at predementia stages with either mild or no cognitive symptoms. However, the design of pre-clinical studies does not follow this attitude, in particular regarding the choice of animal models, often irrelevant to mimic predementia Late Onset Alzheimer’s Disease (LOAD).
Objectives
We aimed to pharmacologically validate the AAV-AD rat model to evaluate preventive treatment of AD.
Methods
We evaluated an N-methyl-D-aspartate receptor antagonist, named memantine, in AAV-AD rats, an age-dependent amyloid rat model which closely mimics Alzheimer’s pathology including asymptomatic and prodromal stages. Memantine was used at a clinically relevant dose (20 mg daily oral administration) from 4 (asymptomatic phase) to 10 (mild cognitive impairment phase) months of age.
Results
A 6-month treatment with memantine promoted a non-amyloidogenic cleavage of APP followed by a decrease in soluble Aβ42. Consequently, both long-term potentiation and cognitive impairments were prevented. By contrast, the levels of hyperphosphorylated endogenous tau remained unchanged, indicating that a long-term memantine treatment is ineffective to restrain the APP processing-induced tauopathy.
Conclusions
Together, our data confirm that relevant models to LOAD, such as the AAV-AD rat, can provide a framework for a better understanding of the disease and accurate assessment of preventive treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) dementia phase (average duration of 8–10 years) is characterized by a gradual alteration of memory, learning abilities and language skills and causes behavioral and personality changes before death (1). Extracellular amyloid plaques and intracellular neurofibrillary tangles (NFTs) are the two main hallmarks of the disease and are mainly composed of amyloid-β peptides (Aβ40 and Aβ42) and hyperphosphorylated tau (P-tau), respectively (2, 3). AD is a chronic illness with long asymptomatic and prodromal phases (duration >20 years) which appear before the dementia phase (3). One of the initial events leading to AD is the accumulation of soluble forms of Aβ and an imbalance between Aβ production and clearance (3). Hyperphosphorylation of tau is suspected to be a downstream process. While pathological concentrations of Aβ are caused by APP, PSEN1 or PSEN2 mutations in Early-Onset Alzheimer’s Disease (EOAD), EOAD patients only represent 1–5% of all AD cases (4) and with a dementia onset before 65 years old. If the etiology of EOAD is well understood, etiology of Late Onset Alzheimer’s Disease (LOAD, patients with a dementia onset > 65) is not. However, some non-congenital etiologies have been recently proposed for LOAD leading to an increased Aβ42 (5): (i) an increase in copy number of the APP gene in a limited subset of neurons (6), (ii) the appearance of neuronal somatic APP mutations known to be associated with familial forms of AD (7) and (iii) viral or bacterial infections (8). The main difference with EOAD is that somatic neo-mutations (mosaicism), APP gene duplications (aneuploidy) or infectious consequences appear obviously only from adulthood. This explains a pre-pathological phase in LOAD that does not exist in EOAD patients. The presence of this phase could thus explain the difference in age of dementia onset between EOAD and LOAD patients.
Thereby, the progression of LOAD can be subdivided into 4 phases: pre-pathological, asymptomatic, prodromal and finally, dementia (Fig. 1). In pre-pathological individuals, cerebral Aβ42 levels are still considered as normal and the disease is not initiated. In asymptomatic individuals when the disease sets in, brain levels of Aβ42 (and potentially P-Tau) slowly begin to increase. At this stage, LOAD individuals are still CSF biomarker negative (9). In the prodromal stage (or mild cognitive impairment due to AD), patients develop the first cognitive symptoms due to amyloid (and potentially P-tau) toxicity (10, 11) and are in the conversion phase for CSF biomarkers. Despite the rise of CSF Aβ and p-TAU starts early, the threshold of positivity for both amyloid and tau markers is certainly reached only in late MCI or dementia in most cases (12). Both asymptomatic and prodromal phases constitute the predementia stage of LOAD where cognitive symptoms are defined by a Clinical Dementia Rating (CDR) score ≤ 0.5 (13). Patients then develop symptoms of dementia. Almost all, if not all, demented patients are positive for CSF biomarkers (A+T+) (12).

AAV-AD rats mimic the LOAD progression
Over the past decade, a major shift in patient inclusion criteria for clinical trials has been observed. In 5 years (2016–2020), the optimal therapeutic window to treat patients with LOAD shifted from the dementia to the predementia phase. Thus, if 60% of the drugs were tested in 2016 in patients with mild to severe dementia, it decreased to 43% in 2020 (14, 15). Consequently, an increase in clinical trials recruiting patients at the prodromal stage was observed (from 31% to 48%). In parallel, at least three phase 2 or 3 clinical trials achieved the primary outcome of significantly slowing cognitive decline (10) and one drug (Adulhelm™ aka aducanumab) has been approved by the Food and Drug Administration (FDA). Despite this evolution in the criteria of patient recruitment, it has not been accompanied by a change in practices in pre-clinical animal studies. Thus, the animal models used to evaluate drugs acting during the predementia stages have remained the same (16).
While AD is an aging disease, in many transgenic mouse models of AD, amyloid plaques and/or NFTs appear very early (17) because of higher concentrations of Aβ and p-Tau compared to what is observed in humans (16). These models try to reproduce the congenital etiology of EOAD (such as Tg2576, APP23, APP/PS1, PDAPP,5xFAD or 3xTg mice) (16, 18) resulting in the absence of pre-pathological phase and symptoms appearance not relevant with the LOAD age range. What was an advantage some time ago, when the aim was to produce amyloid plaques and/or tangles, and cognitive impairments comparable to the dementia phase as quickly as possible to evaluate symptomatic compounds, has become a limitation for evaluating prevention approaches, when brain lesions are only present in a limited number of patients, CSF biomarkers still negative and cognitive impairment still mild (12). The use of animal models mimicking with pertinence the predementia phases of the LOAD is thus needed to evaluate preventive strategies for LOAD patients (19). To overcome the lack of animal models specifically reproducing the predementia phase of LOAD, we developed the AAV-AD rat model (20). Unlike transgenic models, the AAV-AD model is based on the induction of mosaicism and aneuploidy of the APP gene in the hippocampus, thus reproducing a non-congenital etiology close to that of LOAD (6, 7, 20). The gene transfer-based technology used was conducted on 2-month-old adult animals. Therefore, AAV-AD rats presented the age-related progression of LOAD by including its pre-pathological phase.
This study aims to pharmacologically validate the AAV-AD rat model by evaluating preventive therapeutic properties of memantine. Memantine is a N-methyl-D-aspartate receptors (NMDAR) uncompetitive antagonist with moderate affinity that has been approved by the U.S. FDA in 2003 to treat the symptoms from moderate to severe dementia in Alzheimer’s disease. To specifically address this question, we studied the effect of oral treatment with memantine in AAV-AD rats. The treatment lasted 6 months and was started in 4 month-aged rats (equivalent to the asymptomatic phase, defined by a cerebral production of Aβ42 peptides without memory impairment nor LTP defect) and tested at 10 months (equivalent to the prodromal phase, defined by a cerebral production of Aβ42 peptides associated to mild memory impairment and LTP defect) (20, 21) (Fig.1), to replicate an experimental design close to that of a preventive clinical trial (22). We showed that memantine promotes the non-amyloidogenic processing of APP, preventing mild cognitive impairment onset in 10-month-old AAV-AD rats. Additionally, behavioral symptoms (mild anxiety) and tau hyperphosphorylation remained unchanged, suggesting that combining therapies may be required over the long term.
Methods
Animals
Male Wistar rats (eight-week-old; SARL JanvierLabs, Le Genest Saint Isle, France) were used in this study. All experiments were conducted in accordance with the ethical standards of French and European regulations (European Communities Council Directive 2010/63/EU, authorization number APAFIS#4449-2016031012491697).
AAV-AD induction
Rats were anesthetized by an intraperitoneal injection of ketamine/xylazine and placed in a stereotactic frame (Stoelting, Wood Dale, IL, USA). Stereotactic intracerebral injections of AAVs into the hippocampi of both hemispheres were performed, using the following coordinates: antero-posterior: −3.6 mm, lateral: ± 2.5 mm, ventral: −3.3 mm relative to bregma. A volume of 4 µl of viral preparation was injected into each site (2.5×1010 vg/ site and 5×1010 vg/site for PS1 and APP, respectively) at a rate of 0.25 µL/minute. Two groups of animals were produced according to the viral vectors injected or co-injected: AAV-CAG-PS1M146L (control rats), and AAV-CAG-APPSL + AAV-CAG-PS1M146L (AAV-AD rats).
Memantine
Memantine (1-amino-3,5-dimethyl-adamantane) HCl was provided by Carbosynth (Staad, Switzerland) in a purity of 99%. Controls rats and placebo-treated AAV-AD rats were treated for 6 months with vehicle only (water) and memantine-treated AAV-AD rats with a clinically relevant dose of memantine (20 mg/day) after daily water consumption assessment during first week.
Tissue collection and sample preparation
Rats were anesthetized with ketamine/xylazine and were first placed in a stereotactic frame to collect CSF by puncturing the cisterna magna with a low dead space syringe. Brain was dissected for isolation of the hippocampus and the cerebral cortex. Samples were homogenized in a lysis buffer (150 mM NaCl and 1% Triton in Tris-buffered saline) containing phosphatase (Pierce) and protease (Roche) inhibitors and centrifuged for 20 min at 15000 × g.
ELISA
Soluble Aβ was quantified using the V-PLEX Plus Aβ Peptide Panel 1 (6E10) Kit (Meso Scale Diagnostics, Rockville, USA). βCTF was quantified using the IBL APP βCTF Assay Kit (IBL International GmbH, Hamburg, Germany). sAPPα concentrations were assessed using the sAPP-alpha high sensitive ELISA (IBL International GmbH, Hamburg, Germany). Hyperphosphorylated tau was quantified using the Innogenetics Phosphotau 181P kit (Fujirebio Europe, Ghent, Belgium). The ELISA was performed according to the kit manufacturer’s instructions in each case.
Western blotting
Equal amounts of protein (30 µg) were separated by electrophoresis in NuPAGE Bis-Tris Gels (Life Technologies) and transferred to nitrocellulose membranes. The membranes were hybridized with various primary antibodies: APP 6E10 (1/500, Covance), PS1 (1/1000, Millipore), APP C-ter (1/500, Millipore), GAPDH (1/1000, Abcam), P-tau Thr 212 (1/1000, Invitrogen), P-tau Thr 422 (1/1000, Invitrogen), P-CamkII Thr 286 (1/1000, Abcam). Various secondary antibodies were also used: ECL horseradish peroxidase linked anti-rabbit, ECL horseradish peroxidase linked Anti- mouse, ECL horseradish peroxidase linked anti-rat (all 1/2000, GE Healthcare).
Behavioral assessment
Open-field. The apparatus consisted of an open-topped, opaque plexiglas box measuring 90 × 90 × 70 cm placed in a room with controlled dim lighting (40 lux) and constant white noise at 40 dB. Rats were placed in the center of the arena and a video recording was made over a period of five minutes. The behavior of the animals was analyzed from this video. The arena was divided into a central region and a peripheral region, and the time spent in the center and periphery of the open-field was measured.
Morris water maze. Experiments were performed in a tank of 180 cm in diameter and 50 cm deep, filled with opacified water kept at 21°C, and equipped with a platform of 18 cm in diameter, submerged 1 cm below the surface of the water. Visual clues were positioned around the pool and the luminosity was maintained at 350 lux. The rats were initially exposed to a learning phase, which consisted of daily sessions (three trials per session) on five consecutive days. The starting position varied pseudo-randomly, between the four cardinal points. A mean interval of 20 min was left between trials. The trial was considered to have ended when the animal reached the platform. The memorization of the platform position by the animals was validated by a probe test (in which the platform was no longer available) carried out 4 hours after the last training trial to confirm the good memorization of the platform position and thus intact learning capacities, intact learning abilities with a clear spatial bias. Accelerated forgetting was assessed 120 hours after the last training trial in a probe trial in which the platform was no longer available. Animals were monitored using EthoVision software.
Ex vivo electrophysiology
Rats were anesthetized with halothane and decapitated. The brain was rapidly removed from the skull and placed in chilled (0–3°C) artificial cerebrospinal fluid (ACSF) containing 124 mM NaCl, 3.5 mM KCl, 1.5 mM MgSO4, 2.5 mM CaCl2, 26.2 mM NaHCO3, 1.2 mM NaH2PO4, 11 mM glucose. Transverse hippocampal slices (300–400 µm thick) were cut with a vibratome and placed in ACSF in a holding chamber, at 27°C, for at least one hour before recording. Each slice was individually transferred to a submersion-type recording chamber and submerged in ACSF continuously perfused and equilibrated with 95% O2, 5% CO2.
Extracellular recordings were obtained at room temperature from the apical dendritic layer of the CA1 area using glass micropipettes (2-5MQ) filled with 2M NaCl. Presynaptic fiber volleys (PFVs) and AMPAR-mediated field excitatory postsynaptic potentials (fEPSPs) were evoked at 0.1 Hz by electrical stimulation of Schaffer collaterals and commissural fibers located in the stratum radiatum. In addition, specific NMDAR-mediated fEPSPs were isolated in slices perfused with low Mg2+ (0.1mM) containing aCSF and supplemented with the non-NMDA-R antagonist 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzoquinoxaline-7-sulfonamide (NBQX, 10µM). In all cases, the averaged slope of three PFVs and fEPSPs was measured using Win LTP software. To evaluate the level of receptor activation of basal synaptic transmission, the fEPSP/PFV ratio was plotted against stimulus intensity (300, 400 and 500 µA).
For the long-term potentiation (LTP). Theta-burst stimulation (TBS), mimicking the natural stimulation at the theta frequency from the medial septum to the hippocampus, consisted of five trains of four 100 Hz pulses each, separated by 200 ms and delivered at the test intensity. The sequence was repeated three times, with an inter-burst interval of 10 s. Testing with a single pulse was resumed for 60 min (TBS) to determine the level of LTP.
Statistical analysis
Data are expressed as the mean ± SEM. Statistical analyses were performed using GraphPad Prism (GraphPad Soft- ware, La Jolla, CA, USA) and the statistical significance was set to a p-value < 0.05 for all tests. One-way ANOVA followed by Tukey’s post-hoc test or two-way ANOVA followed by Sidak’s post-hoc test were used to determine the significance of differences between groups. Student’s t test was used when only two groups were analyzed.
Results
Memantine does not prevent transgenes expression in AAV-AD rats
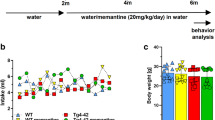
AAV-AD rats were generated using co-injections of AAV vectors encoding human mutant PS1M146L and human mutant APPSL (Swedish & London mutations) genes into the hippocampus (stratum lacunosum moleculare) of two-months-old male Wistar rats. Control rats were only injected with AAV-PS1M146L, as previously described (20). Three groups were used in this study: placebo-treated control rats (n=10), placebo-treated AAV-AD rats (n=10) and memantine-treated AAV-AD rats (n=9). Memantine was delivered through drinking water at a clinically relevant dose (20 mg/day) and given during 6 consecutive months: from 4 (asymptomatic phase) to 10 months (mild cognitive impairment) of age (Fig. 2A). The western blot analysis from whole hippocampi extracts revealed similar protein levels of endogenous APP and PS1 proteins in the 3 groups (Fig. 2B). Both Human APP and cleaved PS1 (30 kDa) levels (20) were similar in both placebo- and memantine-treated AAV-AD rats (Fig. 2B).

Memantine does not modify AAV-AD induction
We bilaterally injected AAV vectors encoding human mutated PS1M146L and APPSL into the stratum lacunosum of rat hippocampi. Rats received either the AAV-CAG-PS1M146L vector (control, n = 10) or both AAV-CAG-PS1M146L and AAV-CAG-APPSL vectors (AAV-AD, n = 20). AAV-AD rats were treated with placebo or memantine (20 mg / kg / day in oral drinking). (A) Schematic representation of the experimental timeline. (B) Western blotting experiments showing the expression of human APP (6E10 antibody), total APP (rat + human forms; APP C-ter antibody) Total PS1 (full length) and Active PS1 (N-terminal fragment), confirming transgene expression 10 months after injection independently of memantine treatment. Bars represent the mean ± SEM. Statistical analyses were performed using one-way ANOVA followed by Tukey’s post-hoc test. Significant differences between placebo-treated control rats and placebo-treated AAV-AD rats are indicated by **p < 0.005. AU: arbitrary units.
Glutamate-mediated neurotoxicity is absent in 10-month-old AAV-AD rats
Memantine, a moderate-affinity NMDA receptor antagonist, is indicated for the treatment of moderate to severe AD to reduce glutamate-mediated excitotoxicity (23). We investigated whether memantine may modulate this excitotoxicity in AAV-AD rats. First, similar protein levels of synaptophysin were observed in all groups (One-way ANOVA, p = 0.768), suggesting no major disturbances of the pre-synaptic compartment. We then assessed the AMPA PFV/Intensity at CA3/ CA1 hippocampal synapses through electrophysiological recordings and confirmed no changes (Two-way ANOVA, Group effect, p = 0.269) (Fig. 3A). In addition, no differences were observed for the post-synaptic marker PSD95 (One-way ANOVA, p = 0.952) as for the AMPA field /Intensity recordings (Two-way ANOVA, Group effect, p = 0.351) (Fig. 3B). Finally, neither AMPA (Two-way ANOVA, Group effect, p = 0.468) or NMDA synaptic activation (Two-way ANOVA, Group effect, p = 0.733) were perturbed in AAV-AD rats compared to controls rats and memantine had no effect on these parameters (Fig. 3C and D). Overall, AAV-AD rats exhibited no signs of glutamate-mediated excitotoxicity at 10 months age and memantine treatment did not exhibit any effect.

Absence of glutamate-mediated neurotoxicity in AAV-AD rats
Synaptic integrity was evaluated in AAV-AD and control rats treated or not with memantine. (A) Pre-synaptic integrity evaluation. Left panel: representative synaptophysin western blot and quantification. Right panel: AMPA PFV/ Intensity assessed by increased stimulation intensities of afferent fibers at hippocampal CA3/CA1 synapses. (B) Post-synaptic integrity evaluation. Left panel: representative PSD-95 western blot. Right panel: AMPA field/Intensity assessed by electrophysiological recordings (C) Ratio AMPA fEPSP/PFV determining basal synaptic activation assessed by electrophysiological recordings. (D) Ratio NMDA fEPSP/PFV representing the isolated NMDA receptor activation assessed by electrophysiological recordings.
Memantine prevents AAV-AD rats mild cognitive impairment
Long term memantine treatment was evaluated on spatial learning and memory using the Morris Water Maze paradigm (20) (Fig. 4A). The learning phase showed no statistical significant differences suggesting that all rats learnt the platform position (Two-way ANOVA, Group effect, p = 0.835; data not shown). The probe test conducted 4 hours after the last day of training confirmed that all groups have normal memory capabilities (Two-way ANOVA, Group effect, p = 0.467; Fig. 4A). By contrast, the probe test processed 120 hours after the last training trial showed that placebo-treated AAV-AD rats traveled in average a lower distance in the target quadrant compared to placebo-treated controls rats (Placebo-treated control: 4.84 ± 0.35 m; placebo-treated AAV-AD: 3.46 ± 0.21 m; two-way ANOVA followed by Tukey’s post-hoc test: p < 0.001) (Fig 4B). Strikingly, memantine-treated AAV-AD rats showed an increase of the distance traveled in the target quadrant compared to placebo-treated AAV-AD rats (Memantine-treated AAV-AD: 4.59 ± 0.25 m; placebo-treated AAV-AD: 3.46 ± 0.21 m; two-way ANOVA followed by Tukey’s post-hoc test: p = 0.005) supporting that memantine avoids accelerated forgetting in AAV-AD rats (Fig. 4B).

Preventive properties of memantine during predementia AD
Memantine was evaluated on behavior, synaptic plasticity, CaMKII phosphorylation and APP processing. (A-B) Probe trial performance at 4h (A) and 120h (B) after the last training session on the Morris water maze task using 10 months old rats. Bottom part represents occupancy plots to visualize the areas in which the animals spent most of the time during the test. (C-D) Long term potentiation (C) were evaluated in placebo-treated controls rats (n=5), placebo-treated AAV-AD rats (n=5) and placebo-treated AAV-AD rats (n=5) while phospho-CaMKII [Thr286] (D) was assessed by western blotting in placebo-treated controls rats (n=5), placebo-treated AAV-AD rats (n=6) and placebo-treated AAV-AD rats (n=7). (E-F) Analysis of cerebral APP product. Soluble Aβ42 (E) assessed by MSD multiplex and evaluation of the Aβ42/ Aβ40 ratio (F). Analysis of β-CTF (G), sAPPα (H) and evaluation of sAPPα/ Aβ42 ratio (I). (J-L) Analysis of CSF amyloid peptides. CSF soluble Aβ40 (J) and Aβ42 (K) assessed by MSD multiplex and evaluation of the CSF Aβ42/ Aβ40 ratio (L). Significant differences between placebo-treated control rats and placebo-treated AAV-AD rats are indicated by *p < 0.05 and ***p < 0.0005. Significant differences between placebo — and memantine - treated AAV-AD rats are indicated by #p < 0.05 and ###p < 0.0005.
Memantine rescues synaptic plasticity in AAV-AD rats
AAV-AD rats treated with placebo (n = 15 slices/N = 5 rats) exhibited impaired LTP magnitude relative to controls rats also treated with placebo (n = 13 slices/N = 5 rats; Placebo-treated control: 128.2 ± 4.80 %; placebo-treated AAV-AD: 110.7 ± 3.54 %; one-way ANOVA followed by Tukey’s post-hoc test, p = 0.038; Fig. 4C). Memantine-treated AAV-AD rats (n = 12 slices/N = 5 rats) exhibited significantly improved LTP (Placebo-treated AAV-AD: 110.7 ± 3.54 %; memantine-treated AAV-AD: 129.4 ± 6.67 %; one-way ANOVA followed by Tukey’s post-hoc test, p = 0.029; Fig. 4C). The calcium-calmodulin-dependent kinase II (CaMKII), in particular its autophosphorylation on Thr286, is essential for long-term potentiation (LTP) and spatial learning in the hippocampus (24–26). Levels of calcium/calmodulin-dependent kinase II (CaMKII) phosphorylation (Thr 286) were decreased in placebo-treated AAV-AD rats compared to controls rats (Placebo-treated control: 1.00 ± 0.02 ; placebo-treated AAV-AD: 0.60 ± 0.07 ; one-way ANOVA followed by Tukey’s post-hoc test, p = 0.054 ; Fig. 4D) (24, 26) whereas memantine treatment prevented this reduction in AAV-AD rats (Placebo-treated AAV-AD: 0.60 ± 0.07; memantine-treated AAV-AD: 1.64 ± 0.12 %; oneway ANOVA followed by Tukey’s post-hoc test, p < 0.001; Fig. 4D).
Memantine decreases soluble Aβ42 levels in AAV-AD rats
Although memantine treatment had no effect on Aβ40 levels (Placebo-treated AAV-AD: 10.61 ± 3.64 pg/mL; memantine-treated AAV-AD: 7.98 ± 3.43 pg/mL ; oneway ANOVA followed by Tukey’s post-hoc test, p = 0.835; data not shown), Aβ42 levels were reduced in the hippocampi of memantine-treated AAV-AD rats (Placebo-treated AAV-AD: 19.43 ± 6.09 pg/mL; memantine-treated AAV-AD: 4.84 ± 2.51 pg/mL; one-way ANOVA followed by Tukey’s post-hoc test, p = 0.048; Fig. 4E). Consequently, memantine decreased the Aβ42/Aβ40 ratio in hippocampi (Placebo-treated AAV-AD: 1.54 ± 0.24; memantine-treated AAV-AD: 0.46 ± 0.14; Student’s t-test, p = 0.017; Fig. 4F). We then evaluated βCTF and sAPPα concentrations and showed that memantine reduced βCTF (Placebo-treated AAV-AD: 11.99 ± 2.18 pmol/L; memantine-treated AAV-AD: 5.69 ± 1.33 pmol/L; oneway ANOVA followed by Tukey’s post-hoc test, p = 0.023 ; Fig. 4G) while increasing sAPPα (Placebo-treated AAV-AD: 0.12 ± 0.07 ng/mL; memantine-treated AAV-AD: 0.48 ± 0.15 ng/mL; one-way ANOVA followed by Tukey’s post-hoc test, p = 0.097 ; Fig. 4H). Memantine rebalanced the sAPPα/Aβ42 ratio in AAV-AD rats (Placebo-treated AAV-AD: 10.08 ± 4.43; memantine-treated AAV-AD: 154.7 ± 50.75; Student’s t-test, p = 0.0219; Fig. 4I). We also evaluated the effect of memantine treatment on CSF Aβ42 concentration. We observed, between placebo- and memantine-treated AAV-AD rats, a trend for increased Aβ40 concentrations and decreased Aβ42 peptides in CSF, though not statistically significant (Fig.4J-K), leading to a reduced Aβ42 /Aβ40 ratio in CSF (Placebo-treated AAV-AD: 0.30 ± 0.04; memantine-treated AAV-AD: 0.11 ± 0.04; Student’s t-test, p = 0.021; Fig.4L).
Memantine does not prevent mild anxiety in AAV-AD rats
All rats travelled similar total distances during the open-field test, demonstrating the absence of motor abnormalities or hyperactivity in each group (One-way ANOVA, p = 0.9867; data not shown). Nevertheless, placebo-treated as well as memantine-treated AAV-AD rats spent less time in the center of the open-field apparatus compared to placebo-treated controls (Placebo-treated control: 3.08 ± 1.04 s; placebo-treated AAV-AD: 0.76 ± 0.38 s; one-way ANOVA followed by Tukey’s post-hoc test: p = 0.068 and memantine-treated AAV-AD: 0.87 ± 0.36 s; one-way ANOVA followed by Tukey’s post-hoc test: p = 0.075) suggesting that memantine did not prevent the mild anxiety behavior (Fig. 5A).

Limitations of memantine as secondary preventive approach in AD
(A) Time in seconds spent in the center of the open-field. Bottom part represents occupancy plots to visualize the areas in which the animals spent the most time during the test. (B -D) Analysis of total tau (B), phospho-tau at [Thr 181] (pTAU 181) (C) and phospho-tau at [Ser 422] (pTAU 422) (D) by ELISA and western blotting respectively. Significant differences between placebo-treated control rats and placebo-treated AAV-AD rats are indicated by *p < 0.05 and **p < 0.005.
Memantine has no effect on endogenous tau pathology
A trend towards increased total endogenous tau concentration was observed (One-way ANOVA, p = 0.072; Fig.5B). We previously described that AAV-AD rats developed increased concentrations of tau phosphorylation level including two epitopes at Thr181 and Ser422 (20). Here, we confirmed that P-Thr181 is increased in AAV-AD rats compared to controls (Placebo-treated control: 51.93 ± 10.20 pg/mL; placebo-treated AAV-AD: 128.50 ± 16.80 pg/mL; one-way ANOVA followed by Tukey’s post-hoc test, p = 0.008; Fig. 5C) and that memantine-treated AAV-AD rat levels remained increased compared to controls (Placebo-treated control: 51.93 ± 10.20 pg/mL; memantine-treated AAV-AD: 127.80 ± 16.42 pg/mL; one-way ANOVA followed by Tukey’s post-hoc test, p = 0.009; Fig. 5C). Similarly, we observed increased levels of P-Ser422 tau in AAV-AD rats compared to controls (Placebo-treated control: 1.00 ± 0.17; placebo-treated AAV-AD: 4.04± 0.80; one-way ANOVA followed by Tukey’s post-hoc test, p = 0.047; Fig. 5D) but not in memantine-treated animals (Placebo-treated control: 1.00 ± 0.17; memantine-treated AAV-AD: 5.04 ± 0.98; one-way ANOVA followed by Tukey’s post-hoc test, p = 0.007; Fig. 5D).
Discussion
The patient inclusion criteria for AD clinical trials are evolving. Additionally, a few phases 2 or 3 trials have reached their endpoints, supporting the idea and the need of an early treatment to delay and/or prevent AD progression before dementia. Nonetheless, practices in the design of preclinical animal studies have not changed, transgenic animal models used mainly mimicking EOAD dementia phase. To evaluate prevention for LOAD patients (19), it is necessary to use animal models that adequately mimic the LOAD predementia phase. To do so, we developed and characterized the AAV-AD rat based on gene transfer (20). In the case of the AAV-AD rat model, control animals are induced by intra-hippocampal injection of the virus encoding the PS1 gene and model animals are induced by intra-hippocampal injection of the virus encoding the PS1 gene and the virus encoding the APP gene. Thus, the AAV-AD model results in increased copy number of the APP gene with Swedish-London mutations compared to control animal, genetic changes restricted to fewer hippocampal neurons. It thus mimics a suspected etiology for LOAD: somatic neo-mutations (mosaicism) (6) and APP gene duplications (aneuploidy) (7) obviously appearing only from adulthood. Soluble Aβ production increases progressively in AAV-AD hippocampi following a slow age-dependent process from the adulthood (equivalent to the asymptomatic phase). Then, this progressive amyloidogenic processing of APP induces hyperphosphorylation of the endogenous tau protein that occurs simultaneously with mild cognitive impairment onset observed with both Morris water maze and LTP recordings (20). Finally, classical hallmarks of AD such as cerebral amyloid angiopathy (CAA), amyloid plaques and neurofibrillary tangles complete the LOAD phenotype in aged rats (30 months after induction) (20). In this study, we show that memantine administrated for 6 months at a clinically relevant dose from the asymptomatic stage (4-month-old rats) prevents the onset of mild cognitive impairment in 10-month-old AAV-AD rats but failed to avoid Tau pathology.
From more than 20 years, memantine is indicated for the treatment of moderate-to-severe dementia in AD patients. In both Europe and United States the recommended dose is between 5 to 20 mg/day (27). Memantine is a moderate-affinity antagonist of NMDAR channels and binds within the ion channel at (or near) the Mg2+ site (28). Although memantine is unable to act on NMDA channels during physiological conditions, it becomes effective in excitotoxicity conditions when the receptors present an extended opening. This may explain its potential neuroprotective effect against excessive glutamate concentration nearby NMDA receptors (29). However, the relevance of memantine as a preventive treatment is still unknown.
As confirmed by the analysis of proteins reflecting the synaptic integrity (Synaptophysin and PSD95) as well as electrophysiological recordings of both pre- and post-synaptic activities, the present study suggests that memantine exerts preventive properties in AAV-AD rats despite the absence of glutamatergic excitotoxicity. Furthermore, we show that memantine (administrated in the drinking water for 6 months) leads to reduced brain Aβ42 and βCTF concentrations. Moreover, full-length APP levels and Aβ40 concentration were unchanged although sAPPα concentration is enhanced. These data suggest that memantine rebalances the APP processing toward the non-amyloidogenic pathway. This could be eventually explained by a lysosomotropic behavior of memantine. Like other amantadine derivatives, memantine is a cationic amphiphilic drug with a structure very similar to other lysosomotropic drugs that accumulate in acidic cellular compartments and inhibit phospholipases. Thus, memantine could reduce the secretion of APP and Aβ peptides through its accumulation in lysosomes (30). In addition, the reduction of Aβ42 is associated with a delayed onset of mild cognitive impairment and supported by rescued LTP. A molecular link between Aβ42 and both LTP and cognition defects has been described as the phosphorylation of CaMKII on the Thr286 epitope (31, 30). We thus confirm in this study this hypothesis since memantine prevents the reduction of Thr286 P-CaMKII, in association with the prevention of both LTP defect and mild cognitive impairment onset in AAV-AD rats. Memantine has already been described in different animal models. These different preclinical studies conclude that memantine has no pro-memory effect outside the pathological context with no behavioral effects of memantine on control animals (32–34). This suggests that memantine action mode leading to cognitive beneficial effect observed in memantine-treated AAV-AD rats is specific to Alzheimer’s disease. Memantine has been also tested in AD transgenic mice such as APP/PS1 (33), 3xTg-AD (32) or Tg4-42 mouse model (34). However, these mice exhibit severe cognitive impairment in the Morris water maze, which cannot be considered as mild cognitive impairment (21) restricting their use to assess preventive aspect for drug preclinical evaluation. Thereby, if transgenic mice can be useful tools to characterize the cognitive effect of symptomatic treatments (in particular for EOAD forms), the extremely fast progression of the disease in these models make them less accurate to characterize primary or secondary preventive treatments. Their use, in association with the AAV-AD model, remains nevertheless essential for a complete preclinical characterization of drugs to evaluate both preventive and curative properties. For example, the results described in this study are consistent with and complementary to those observed in memantine-treated Tg4-42 mice (34) that express N-truncated human Aβ4-42 without any other mutation (35). By combining the results of the two studies, it is thus possible to obtain a complete picture of the effect of memantine on the amyloid component when administered as secondary prevention.
Lastly, we observed that memantine did not prevent tau hyperphosphorylation in AAV-AD rats where phosphorylation on Thr181 and Ser422 epitopes remained high and similar to AAV-AD rats treated with placebo. Strikingly, memantine treatment in 3xTg-AD mice was associated with a decline in the levels of hyperphosphorylated tau and cortical tangles density. Similarly to the behavioral impairments, 3xTg-AD mice show very rapid tangles formation (36) which does not seem compatible with a predementia context as in human.
Altogether, our data provide information about memantine benefits and limitations as a secondary prevention of LOAD. While memantine lowers amyloid load and prevents mild cognitive onset, it does not abolish tau hyperphosphorylation in the AAV-AD rat model, raising questions about the effect of memantine over longer periods of time, especially on its impact on tangle formation. In a clinical landscape where the FDA recently approved aducanumab for the treatment of AD from prodromal stage, more and more trials will aim to include patients before the dementia phase and evaluate disease-modifying drugs. This is not in line with most animal models — originally designed to model AD dementia - currently used. Fortunately, the limitations of these models raised in the current manuscript are known by the scientific community and several labs and consortium (such as www.model-ad.org) are already working on the new generation of improved and more accurate LOAD models.
References
Honig LS, Mayeux R. Natural history of Alzheimer’s disease. Aging (Milano). 2001;13(3):171–82. https://doi.org/10.1007/BF03351476.
Perl DP. Neuropathology of Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):32–42. https://doi.org/10.1002/msj.20157.
Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1:15056. https://doi.org/10.1038/nrdp.2015.56.
Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther. 2011;3(1):1. https://doi.org/10.1186/alzrt59.
Morris GP, Clark IA, Vissel B. Questions concerning the role of amyloid-beta in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018;136(5):663–89. https://doi.org/10.1007/s00401-018-1918-8.
Bushman DM, Kaeser GE, Siddoway B, et al. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. Elife. 2015;4. https://doi.org/10.7554/eLife.05116.
Lee MH, Siddoway B, Kaeser GE, et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature. 2018;563(7733):639–45. https://doi.org/10.1038/s41586-018-0718-6.
Sochocka M, Zwolinska K, Leszek J. The Infectious Etiology of Alzheimer’s Disease. Curr Neuropharmacol. 2017;15(7):996–1009. https://doi.org/10.2174/1570159X15666170313122937.
Lafirdeen ASM, Cognat E, Sabia S, et al. Biomarker profiles of Alzheimer’s disease and dynamic of the association between cerebrospinal fluid levels of beta-amyloid peptide and tau. PLoS One. 2019;14(5):e0217026. https://doi.org/10.1371/journal.pone.0217026.
Tolar M, Abushakra S, Hey JA, Porsteinsson A, Sabbagh M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res Ther. 2020;12(1):95. https://doi.org/10.1186/s13195-020-00663-w.
Petersen RC, Caracciolo B, Brayne C, Gauthier S, Jelic V, Fratiglioni L. Mild cognitive impairment: a concept in evolution. J Intern Med. 2014;275(3):214–28. https://doi.org/10.1111/joim.12190.
Allegri RF, Chrem Mendez P, Calandri I, et al. Prognostic value of ATN Alzheimer biomarkers: 60-month follow-up results from the Argentine Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement (Amst). 2020;12(1):e12026. https://doi.org/10.1002/dad2.12026.
Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–4. https://doi.org/10.1212/wnl.43.11.2412-a.
Cummings J, Morstorf T, Lee G. Alzheimer’s drug-development pipeline: 2016. Alzheimers Dement (N Y). 2016;2(4):222–32. https://doi.org/10.1016/j.trci.2016.07.001.
Cummings J, Lee G, Zhong K, Fonseca J, Taghva K. Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement (N Y). 2021;7(1):e12179. https://doi.org/10.1002/trc2.12179.
Nakai T, Yamada K, Mizoguchi H. Alzheimer’s Disease Animal Models: Elucidation of Biomarkers and Therapeutic Approaches for Cognitive Impairment. Int J Mol Sci. 2021;22(11). https://doi.org/10.3390/ijms22115549.
King A. The search for better animal models of Alzheimer’s disease. Nature. 2018;559(7715):S13–S5. https://doi.org/10.1038/d41586-018-05722-9.
Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115(1):5–38. https://doi.org/10.1007/s00401-007-0312-8.
Preuss C, Pandey R, Piazza E, et al. A novel systems biology approach to evaluate mouse models of late-onset Alzheimer’s disease. Mol Neurodegener. 2020;15(1):67. https://doi.org/10.1186/s13024-020-00412-5.
Audrain M, Souchet B, Alves S, Fol R, Viode A, Haddjeri A, et al. betaAPP Processing Drives Gradual Tau Pathology in an Age-Dependent Amyloid Rat Model of Alzheimer’s Disease. Cereb Cortex. 2018;28(11):3976–93. https://doi.org/10.1093/cercor/bhx260.
Li D, Huang Y, Cheng B, Su J, Zhou WX, Zhang YX. Streptozotocin Induces Mild Cognitive Impairment at Appropriate Doses in Mice as Determined by Long-Term Potentiation and the Morris Water Maze. J Alzheimers Dis. 2016;54(1):89–98. https://doi.org/10.3233/JAD-150979.
Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6(228):228fs13. https://doi.org/10.1126/scitranslmed.3007941.
Molinuevo JL, Llado A, Rami L. Memantine: targeting glutamate excitotoxicity in Alzheimer’s disease and other dementias. Am J Alzheimers Dis Other Demen. 2005;20(2):77–85. https://doi.org/10.1177/153331750502000206.
Fukunaga K, Muller D, Miyamoto E. CaM kinase II in long-term potentiation. Neurochem Int. 1996;28(4):343–58. https://doi.org/10.1016/0197-0186(95)00097-6.
Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279(5352):870–3. https://doi.org/10.1126/science.279.5352.870.
Lledo PM, Hjelmstad GO, Mukherji S, Soderling TR, Malenka RC, Nicoll RA. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci U S A. 1995;92(24):11175–9. https://doi.org/10.1073/pnas.92.24.11175.
van Marum RJ. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr Dis Treat. 2009;5:237–47. https://doi.org/10.2147/ndt.s4048.
Chen HS, Lipton SA. The chemical biology of clinically tolerated NMDA receptor antagonists. J Neurochem. 2006;97(6):1611–26. https://doi.org/10.1111/j.1471-4159.2006.03991.x.
Danysz W, Parsons CG, Mobius HJ, Stoffler A, Quack G. Neuroprotective and symptomatological action of memantine relevant for Alzheimer’s disease—a unified glutamatergic hypothesis on the mechanism of action. Neurotox Res. 2000;2(2–3):85–97. https://doi.org/10.1007/BF03033787.
Hostetler KY, Richman DD. Studies on the mechanism of phospholipid storage induced by amantadine and chloroquine in Madin Darby canine kidney cells. Biochem Pharmacol. 1982;31(23):3795–9. https://doi.org/10.1016/0006-2952(82)90295-7.
Reese LC, Laezza F, Woltjer R, Taglialatela G. Dysregulated phosphorylation of Ca(2+) /calmodulin-dependent protein kinase II-alpha in the hippocampus of subjects with mild cognitive impairment and Alzheimer’s disease. J Neurochem. 2011;119(4):791–804. https://doi.org/10.1111/j.1471-4159.2011.07447.x.
Martinez-Coria H, Green KN, Billings LM, et al. Memantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic mice. Am J Pathol. 2010;176(2):870–80. https://doi.org/10.2353/ajpath.2010.090452.
Minkeviciene R, Banerjee P, Tanila H. Memantine improves spatial learning in a transgenic mouse model of Alzheimer’s disease. J Pharmacol Exp Ther. 2004;311(2):677–82. https://doi.org/10.1124/jpet.104.071027.
Stazi M, Wirths O. Chronic Memantine Treatment Ameliorates Behavioral Deficits, Neuron Loss, and Impaired Neurogenesis in a Model of Alzheimer’s Disease. Mol Neurobiol. 2021;58(1):204–16. https://doi.org/10.1007/s12035-020-02120-z.
Bouter Y, Dietrich K, Wittnam JL, et al. N-truncated amyloid beta (Abeta) 4–42 forms stable aggregates and induces acute and long-lasting behavioral deficits. Acta Neuropathol. 2013;126(2):189–205. https://doi.org/10.1007/s00401-013-1129-2.
Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–21. https://doi.org/10.1016/s0896-6273(03)00434-3.
Acknowledgement
We thank Noëlle Dufour, and Gwenaëlle Auregan for their help in producing the viral vectors, Gaëlle Dufayet-Chaffaud for her help in the biochemical analyses, and Karine Cambon for advice on animal behaviour.
Funding
Funding: This work was supported by DIM Biotherapies, the Région Ile-de-France, IDEX Paris Saclay, INSERM Transfert, CEA “Technologie Pour la Santé” and by a grant from “Investissement d’Avenir—ANR-11-INBS-0011”—NeurATRIS: A Translational Research Infrastructure for Biotherapies in Neurosciences.
Author information
Authors and Affiliations
Contributions
B.S., M.A., and J.B. carried out the design of the study and wrote the manuscript. B.S., M.A., and J.B. performed the biochemical, behavioral, and statistical analyses. R.F., S.A., N.S.O., S.T. participated in the biochemical analyses and helped to draft the manuscript. P.D., B.P., and J.-M.B. performed the electro-physiological recordings and helped to design the study and draft the manuscript. N.C. read the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
All experiments were conducted in accordance with the ethical standards of French and European regulations (European Communities Council Directive 2010/63/EU, authorization number APAFIS#4449-2016031012491697).
Additional information
Conflict of interest
J.B. is co-founder of AgenT SAS. J.B. and B.S. are employees of AgenT SAS.
Rights and permissions
About this article

Cite this article
Souchet, B., Audrain, M., Alves, S. et al. Evaluation of Memantine in AAV-AD Rat: A Model of Late-Onset Alzheimer’s Disease Predementia. J Prev Alzheimers Dis 9, 338–347 (2022). https://doi.org/10.14283/jpad.2021.67
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.14283/jpad.2021.67