
Abstract
Lovastatin (Lov) is a lipid-lowering agent, with 5% bioavailability (BA) due to extensive first pass metabolism and poor solubility. To enhance dissolution and in vivo effects, Lov solid self microemulsifying drug delivery system (SMEDDS) and liquisolid systems were developed and evaluated to select superior one. Solubilities were determined in oils, surfactants, and cosurfactants. Ternary phase diagrams were constructed and selected the one which showed maximum emulsion zone. In vitro dissolution, DSC, SEM and PXRD studies were used to characterize the developed formulations. In vivo studies were conducted on optimal formulations in wistar rats. Based on solubilities, Capmul PG8 and Capmul MCM were preferred as oils, Labrasol and Transcutol P as surfactant and cosurfactant. Here, Syloid XDP carrier showed better adsorption capacity among others, hence was used in optimal solid SMEDDS (SX) and liquisolid (LS) formulations. Dissolution study results showed significant improvement in release when compared to pure drug. DSC, SEM, and PXRD results indicated the loss of drug crystallinity in optimal formulations. In pharmacokinetic (PK) study, SX and LS showed 2.57 and 1.43 fold improvements in AUC, when compared to that of coarse suspension (CS). In pharmacodynamic (PD) study, hyperlipidemia was induced by Triton X-100. CS and LS treatments showed a decline in hyperlipidemic levels at 4 h. But, SX-treated group showed early onset of decline at 2 h. Further, the duration of anti-hyperlipidemia was at least 12 h extra when compared to CS and LS. This study confirmed the superiority of SX over LS in PK and PD effects.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Oral drug delivery is a kind of noninvasive technique for which no skill is needed for dose intake (1). The investigation of new drug molecules by implementing combinatorial chemistry and high-throughput screening increases both molecular weight and lipophilicity, which finally leads to poor water solubility (2). Nearly 40–70% of the newly developed molecules have a constraint of oral absorption due to their poor dissolution characteristics (3). To improve the dissolution of lipophilic compounds numerous technologies were developed, such as solid dispersions (4), liquisolid compact (5), milling technique (6), nanocrystals (7) and lipid-based delivery systems (8, 9).
In the lipid based delivery systems, SMEDDS (self-microemulsifying drug delivery systems) gained much importance for increasing the dissolution and bioavailability. SMEDDS are isotropic mixtures containing oil, surfactant, and cosurfactant. These have capacity to form oil in water (O/W) emulsion, upon mixing with aqueous medium or gastrointestinal content (10). The lipids and surfactants improve the drug solubilization and absorption by enhancing transcellular and paracellular permeability, and the digested lipids reassemble in enterocyte to stimulate chylomicron production, which favors lymphatic uptake (11, 12).
Lymphatic uptake of lipophilic drugs, using lipid-based systems, is a prominent strategy, for avoiding the first-pass metabolism. The fundamental feature of oral administration is absorption of drug molecules from intestine (13). Effective permeability (Peff) across the intestinal mucosa determines the drug absorption rate and extent (14). Liquisolid formulation is also a dissolution enhancement technique for poor water-soluble drugs or high lipophilic drugs to improve oral bioavailability (15). Liquisolid systems are the formulations developed by converting the liquid drugs, i.e., drug suspensions or drug solutions in to nonadherent, free flowing, dry powders using a carrier along with coating material (16). The improved solubility of liquisolid delivery system is due to its enhanced surface area and coupled with increased wettability of drug particles (17). Self-emulsifying drug delivery systems as liquid formulations have some disadvantages such as drug leakage and few choices of dosage forms (18). To overcome these limitations, liquid self-emulsifying systems can be transformed in to solid dosage forms by using different methods (adsorption on to solid carrier, melt granulation, spray drying, melt extrusion, or nanoparticle formation). Solid self-emulsifying drug delivery systems combine the advantages of liquid lipid formulations with those of solid dosage forms, such as higher stability and longer period of storage.
Lovastatin (Lov) belongs to BCS class II. This is a drug with low dose and exhibits low solubility and high permeability (19). It is a statin category drug and is currently prescribed. The oral bioavailability (BA) of Lov is < 5% due to its poor water solubility and high metabolism by CYP3A4 (20). Lov is a cholesterol-lowering agent used in the treatment of hyperlipidemia. The mechanism of action is by inhibition of HMG Co-A reductase, a key enzyme responsible for the cholesterol biosynthesis (21). Further, the drug Lov reduces risk of heart diseases and prevent strokes and heart attacks. Lov tablets are available in market with different brand names (Lostatin®, lovacard®, Astatin®, Mevacor®).
To improve the BA, many delivery systems of Lov were developed and studied. They being, nanostructured lipid carriers (22), lipid nanoparticles (solid) (23), solid dispersions (24), liquisolid compacts (25), and SMEDDS (26,27,28). In general, liquisolid and solid SMEDDS enhance the dissolution rate of poorly soluble/insoluble drugs, and consequently, contribute for enhanced BA. However, no pharmacokinetic studies were reported for liquisolid formulation and solid SMEDDS of Lov in rats. In addition, no comparative studies were reported till now on Lov solid SMEDDS and liquisolid compacts. The main idea for comparison of SMEDDS and liquisolid formulations is that both the systems are known to improve the dissolution and bioavailability of poorly soluble drugs. But, the bulkiness of liquisolid formulations is more when compared to that of solid SMEDDS for any fixed dose. Consequently, oral administration of bulky dosage form may cause discomfort to patient. Hence, the solid SMEDDS are preferred to liquisolids. Previously, iloperidone (29) and fluvastatin (30) solid SMEDDS were prepared and compared with that of liquisolid formulations of same drug for the extent of dissolution, bioavailability, and pharmacodynamic effects. In both cases, solid SMEDDS of drugs improved oral bioavailability over liquisolid drugs. But, in case of pharmacodynamic effects, iloperidone liquisolids behaved similar to that of solid SMEDDS. In contrast, solid SMEDDS of fluvastatin produced enhanced pharmacodynamic effects when compared to that of liquisolids. Thus, there existed scope for differential effects of liquisolid and solid SMEDDS formulation of same drug in the context of pharmacodynamic effects. Hence, no generalization was envisaged. In liquisolid technique, the drug is solubilized in a suitable nonvolatile solvent and converted in to solid form using a carrier and coating material. SMEDDS is a thermodynamically stable system which forms fine oil globules, contributing for large surface area by self-emulsification in gastrointestinal tract. Consequently, these developed systems could improve the dissolution of poorly soluble/insoluble drugs. But, the drug from SMEDDS has an additional advantage of lymphatic transport due to the formation of chylomicrons. So, this comparative study of these two delivery systems was undertaken to evaluate the relative improvement in dissolution and in vivo drug effects, i.e., pharmacokinetic and pharmacodynamic assessment. Thus, this study is important in selecting a better delivery system for industrial manufacture.
Initially, lovastatin solid SMEDDS and next liquisolid formulation were developed. The transformation of liquid formulations in to free flowing powders was achieved by using suitable porous silicious carriers. Short term stability was also checked. The developed liquisolid formulations and solid SMEDDS formulations were characterized by techniques like SEM (scanning electron microscopy), PXRD (powder X-ray diffractometry), and DSC (differential scanning calorimetry). Further, the optimized liquisolid formulation and solid SMEDDS were evaluated for pharmacokinetic (PK) and pharmacodynamic (PD) effects in male Wistar rats (n = 6).
MATERIALS AND METHODS
Materials
Lovastatin was purchased from M/s Yarrow chemicals Pvt Ltd, Mumbai. Peceol, Lauroglycol 90, Labrasol, Labrafac WL 1349, Labrafil M 2125 CS, Labrafil M 1944 CS, and Transcutol P were obtained as free samples from Gattefosse, USA. Capmul MCM, Captex 355 Capmul PG8, and Capmul PG 12 were supplied by M/s Abitec corporation. Syloid 244 FP and Syloid XDP were free samples obtained from Grace and Co, Mumbai. Neusilin US2 and Fujicalin SG were gift samples from Gangwal Chemicals Pvt. Ltd, Mumbai. Tween 80, PEG 600, PEG 400, and PEG 200 were purchased from Merck, India. Further, remaining chemicals used in this study belong to analytical category, and the solvents employed were of HPLC grade.
Methods
Solubility Studies of Lovastatin
Lov solubility was determined in different vehicles, such as oils, cosurfactants, and surfactants. About 1 g of each vehicle was transferred in to the capped (screw) vial to which excess amount of the Lov was added. The experiment was done in triplicate, and the drugloaded vehicle was subjected to cylomixing using a cyclomixer for 5 min. Then, the resulted mixture was agitated for 48 h on an orbital shaker at 180 rpm (31). Then, vials were centrifuged at 5000 rpm for 10 min to settle the insoluble drug. Then, the clear supernatant phase was separated and passed through 0.45 µm syringe filter (32). Necessary dilutions were made and the amount of drug solublized was estimated using HPLC method (33).
Construction of the Pseudoternary Phase Diagrams
Based on the results of solubility studies, the vehicles showing maximum drug solubility were selected. Using water titration method, pseudoternary phase diagrams were drawn to show the emulsion zones. Further, mixtures of surfactant and cosurfactant (Smix) were prepared in the ratios of 1:1 and 2:1. Then, mixtures of oil and Smix in the ratios of 1:9 to 9:1 were prepared, each having a weight of 1 g. Finally, the mixtures were mixed using a cyclomixer and water titration (dropwise addition) was carried out under gentle agitation to observe the turbidity. The extent of water content needed was found in each case. TriPlot software, USA (version 4.1.2), was used to construct pseudoternary phase diagrams, in which each corner represented 100% of its component (34).
Preparation and Characterization of Liquid SMEDDS
Preparation of the Lov Liquid SMEDDS
The liquid SMEDDS formulations were developed with variable amounts of oil, surfactant, and cosurfactant. Compositions are given in Table II. Ten milligrams of Lov was transferred in to small vial having oil, surfactant and cosurfactant mixture. The mixture was cyclomixed by a vortex mixer for 20 min to form an isotropic mixture. Based on pseudoternary phase diagrams, Smix containing surfactant:cosurfactant in the ratio of 2:1 was selected for preparing formulations, which showed maximum emulsion zone.
Characterization of the Lov Liquid SMEDDS
Measurement of Globular Characters
The size, PDI and zeta potential of the developed formulations were analyzed by Malvern Zetasizer (ZS 90) instrument. The principle involved was photon correlation spectroscopy. Measurement was performed at 25 °C while keeping the angle of detection at 90° (35). About 100 µL of prepared liquid SMEDDS was made to 5 mL (1:50 dilution) with double-distilled water and checked the size, PDI, and zeta potential.
Preparation and Characterization of Lov Solid SMEDDS and Liquisolid Formulations
Preparation of Solid SMEDDS
Optimized Lov liquid SMEDDS was transformed in to solid SMEDDS with four oiladsorbing porous carriers like Neusilin US2, Syloid 244FP, Syloid XDP and Fujicalin SG. The liquid SMEDDS formulation was gradually added to the carrier and mixed in a mortar until a dry and free flowing powder was formed (29).
Preparation of Liquisolid Formulations
Solubility was determined in various nonvolatile solvents like propylene glycol, Transcutol P, Labrasol, Tween 80 and polyethylene glycols like PEG 200, PEG 400 and PEG 600. Results of solubility study revealed the solvent which exhibited maximum solubility of Lov, and it was chosen for the development of liquisolid formulation. The drug containing solvent was added individually to the carriers like Neusilin US2, Avicel PH 102, Syloid XDP and Fujicalin SG in a mortar and mixed. Finally, Aerosil 200 as a coating material was added to form nonadherent, dry, and free-flowing powder. For preparing the liquisolid formulations, the carrier and coating material were used in the ratio of 10:1.
Characterization of Solid SMEDDS and Liquisolid Formulations of Lov
Determination of Liquid Retention Potential, Liquid Load Factor, and Powder Characteristics
Liquid retention potential was defined as the maximum quantity of liquid that was retained on a solid carrier, without compromising the flow behavior at 33° angle of slide (36). The amount of the liquid formulation to be retained by the carrier and coating material is dependent on excipient ratio (R) (37). Excipient ratio is a ratio of weights of the carrier (Q) to that of the coating material (q). The flow character and compressibility of powder would be maintained below the maximum retaining capacity of carrier. The amount of liquid formulation to be retained by unit weight of the carrier is called as liquid load factor (Lf). Liquid load factor is the ratio of weight of the liquid formulation (W) and that of the carrier (Q) and is denoted by the following formula (38),
Among powder flow characteristics, the angle of repose was found by the commonly used fixed funnel method (39). Hausner’s ratio and Carr’s index were calculated by using bulk and tapped densities (40).
Size Analysis of Solid SMEDDS
Here, the total optimized solid SMEDDS formulation (685 mg) was added to 10 ml of double-distilled water and mixed. From this, nearly 100 µl of the suspension was taken, further diluted to 5 ml (1:50 dilution) and determined the particle size, PDI, and zeta potential as per the method used for liquid SMEDDS.
Solid-State Characterization
Drug-Excipient Compatibility Studies by DSC
DSC is a thermoanalytical tool meant for assessment of heat energy uptake by the sample with increase or decrease in temperature (41). In DSC study, heating was applied on the sample and reference cells. Temperature of both cells was gradually increased. The difference in input energy that was needed to match the temperature of the sample to that of the reference is nothing but the heat absorbed or released by the sample (42). DSC study was conducted by using Perkin Elmer DSC 4000 model. Aluminum crucibles were used for loading the samples and nitrogen was used as the purging gas. Approximately 10 mg of the sample was transferred in to a crucible, and the lid was placed and crimped. The study was conducted in temperature range of 20–250 °C with heating at a rate of 10 °C/min to obtain thermograms.
SEM for Studying Surface Morphology
Scanning electron microscope (SEM S-3700, Hitachi, Japan) was used for studying the morphological features of pure drug (lovastatin), optimized solid SMEDDS, and liquisolid formulation. Each sample was fixed on a brass stub and then gold coated as thin layer to make electrically conductive (43). SEM images were obtained at × 60, × 100 and × 250 magnifications.
Characterization of Lov Crystallinity by PXRD
In PXRD studies, nickel filtered CuKα radiation (30 mA and 40 kV) was used and the samples were run at 2-theta (Ɵ) degrees ranging from 2° to 70° at a step angle and step time of 0.045° and 0.5 s, respectively (44). PXRD instrument (Miniflex 600, Rigaku, Japan) was used in this study.
In vitro Dissolution Studies
The dissolution studies of the formulations were conducted in USP type II apparatus (Electrolab, TDT-08L). Dissolution test was performed on pure drug (lovastatin), solid SMEDDS, and liquisolid formulations with 10 mg of lovastatin. Dissolution study was conducted at 37 ± 0.5 °C using 500 ml of 0.1 N HCl as medium with 50 rpm (45). To maintain the sink condition, medium was replaced after removing the sample at regular time intervals (46). Quantification of drug was done by HPLC method (33) and the studies were in triplicate (n = 3).
Pharmacokinetic Study
Bioavailability study was conducted in male Wistar rats, weighing 230–250 g. This study is a parallel, single oral dose and 3 group study. The animals were maintained for overnight in the fasting condition before the study. Animals were grouped in to three and with each group containing six animals. Coarse suspension, optimized solid SMEDDS and optimized liquisolid formulations were given to the three groups. The oral dose at 3.5 mg/kg body weight was given to each rat. This study was approved by the Institutional Animal Ethical committee of UCPSc, Kakatiya University, Warangal, Telangana, India (IAEC/05/UCPSc/KU/2019). Blood samples were withdrawn at following time intervals of 0, 0.5, 1, 2, 4, 6, 8, 12 and 24 h from the rat by puncturing retro orbital venous plexus (47). The tubes were centrifuged for 20 min at 5000 rpm to separate the serum. The collected serum samples were stored at − 20 °C.
Extraction of Lovastatin from Serum Samples and Analysis by HPLC Method
HPLC analysis of Lov was performed as per published method (33) using C18 column (Lichrospher, 250 × 4.6 mm, 5 µm). Acetonitrile and 0.05 M KH2PO4 in the ratio of (70:30) was prepared and used as mobile phase for elution. The phosphate buffer pH was adjusted to 7 by triethanolamine. Flow rate (1 ml/min) was maintained during the elution. Lovastatin was analyzed at λmax 238 nm (33). Liquid–liquid extraction principle was involved in the extraction of lovastatin from serum samples (33). To 100 µl of serum sample, 100 µl of iloperidone (2 µg/ml) solution was added as internal standard. Then, to precipitate the proteins, 100 µl of methanol was added as precipitating agent. At this stage, to extract the drug, 1 ml of diethyl ether was added as an extraction solvent. This mixture was vortexed and centrifuged at 5000 rpm for 10 min. The resulted clear supernatant organic layer containing drug was separated and evaporated to drynesss. Next, 100 µl of the mobile phase was added to dried extract and vortexed. Then, the sample of 20 µl was injected in to the HPLC system for quantification.
Calculation of Pharmacokinetic Parameters and Statistical Comparison
The PK parameters like, AUC, Cmax, Tmax, t1/2 and MRT were obtained from serum Lov concentration vs time profiles, with help of Kinetica 2000 software (version 5.0). AUC indicated the drug bioavailability. All the PK parameters were expressed as mean ± SD. GraphPad Prism software (version 5.03) was used to perform one-way ANOVA (P < 0.05) for statistical comparison of data among the three groups.
PD Study
Pharmacodynamic, i.e., anti-hyperlipidemic activity study was done in male Wistar rats. Animals, in the weight range of 230–250 g, were selected and made into 5 groups. Animals were maintained for overnight in fasting state before the study. Stock solution containing 100 mg/ml of Triton X-100 was made by dissolving 4 g in 40 ml of 0.9% normal saline solution. An intraperitonial injection (i.p.) of Triton X-100, at a dose of 100 mg/kg body, was given to induce hyperlipidemia in male Wistar rats (48, 49).
Animals were grouped into five groups, and each containing six rats.
-
Group A: Normal control
-
Group B: Hyperlipidemic control—Triton X-100 intraperitonially treated group (100 mg/kg)
-
Group C: Lovastatin coarse suspension (3.5 mg/kg) and Triton X-100-treated group
-
Group D: Lovastatin liquisolid (LS) formulation (3.5 mg/kg) and Triton X-100-treated group
-
Group E: Lovastatin solid SMEDDS (SX) (3.5 mg/kg) and Triton X-100-treated group
Drug formulations were administered by oral route after 24 h of Triton X-100 treatment to the animals. Blood was collected from animals at predetermined time points (0, 12, 24, 26, 28, 30, 32, 36, 48, 72 and 96 h) from the retro orbital plexus puncture. Serum was separated from clotted blood by centrifugation. The serum samples were analyzed for lipids like total cholesterol, triglycerides, HDL, LDL and VLDL in all the groups using in vitro diagnostic kits (Tulip diagnostics, Pvt Ltd) and a semi auto analyzer (Erba Chem-7, Germany).
Preparation of Lovastatin Coarse Suspension
Lov coarse suspension was made with sodium carboxymethyl cellulose (NaCMC) as suspending agent. About 50 mg of NaCMC was added to 5 ml of double distilled water in a glass mortar and triturated. Then, 10 mg of lovastatin was added and mixed for 5 min. Finally, the 10 ml volume was made using doubledistilled water.
Stability Studies of Lov Formulations During Short Storage Period
Stability studies were conducted on the optimal solid SMEDDS (SX) and liquisolid (LS) formulations after storing for 90 days at RT (room temperature) (25 °C). The formulations were evaluated for particle size, PDI, zeta potential and drug content (36). Formulations were tested on 1st, 30th, 60th and 90th days.
RESULTS AND DISCUSSION
Solubility Studies
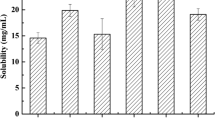
Solubility of lovastatin was found in different vehicles, such as oils, surfactants and cosurfactants in triplicate. In all eight oils, three surfactants and five cosurfactants were included in this study (Table I). Based on relative extent of drug solubility, Capmul PG8 and Capmul MCM as oils, Labrasol as surfactant, and Transcutol P as cosurfactant were chosen in this study. Capmul MCM and Capmul PG8 showed the solubility of 17.56 ± 0.09 mg/g and 36.15 ± 0.21 mg/g, respectively. Labrasol exhibited the solubility of 25.08 ± 0.08 mg/g and Transcutol P showed the solubility of 25.39 ± 0.04 mg/g.
Construction of Pseudoternary Phase Diagrams
Pseudoternary phase plots were drawn to know the emulsion zone, microemulsion, and nonemulsion zone areas. In this study, we obtained the globules falling in the microemulsion zone, not in nanoemulsion range (Fig. 1). Here, oil, Smix (mixture of cosurfactant and surfactant) and water represented the three corners of the phase diagram. Oils (Capmul MCM and Capmul PG8), surfactant (Labrasol) and cosurfactant (Transcutol P) were chosen. Two series of plots were constructed in which series A contained Capmul PG8 and series B contained of Capmul MCM as oils. Surfactant and cosurfactant mixtures in the ratio of 1:1 and 2:1 were made. Then, oil and Smix in the ratios of 1:9 to 9:1 were made and gradually titrated with distilled water to observe the occurrence of turbidity. The percentages of oil, Smix and water required to form an emulsion were identified. These obtained percentage values were incorporated in to TriPlot software to obtain ternary phase diagrams to demarcate emulsion zone and nonemulsion zone. Based on emulsion zone from ternary phase plots, different formulations were prepared within emulsion zone by varying percentages of oil and Smix and evaluated for globular size to identify microemulsion zone (30). Here, Smix ratio of 2:1 showed more emulsion area in both cases of A and B series. Hence, the Smix with a ratio of (2:1) was selected to formulate liquid SMEDDS of lovastatin.

Construction of pseudoternary phase diagrams. Smix contains Labrasol and transcutol P. Series A contains Capmul PG8 and Smix ratios of 1:1 and 2:1. Series B contains Capmul MCM and Smix ratios of 1:1 and 2:1
Preparation and Characterization of Liquid SMEDDS
Preparation of Liquid SMEDDS
A and B series of the liquid SMEDDS were prepared. In “A” series, Capmul PG8 and in B series, Capmul MCM were used as oils. In both the series, Labrasol and transcutol P were included as surfactant and cosurfactant. Formulations, A1, A2, A3 and A4 were made with Capmul PG8 as oil by taking 15%, 20%, 30% and 40% w/w, respectively. In B1, B2, B3 and B4 formulations, Capmul MCM was employed as oil at 15%, 20%, 30% and 40% w/w concentrations respectively (Table II) along with other ingredients.
Characterization of Prepared Liquid SMEDDS
Determination of Globule Size, PDI and ZP
Globule size, PDI and ZP were analyzed for the formulations of both series using a Malvern Zetasizer (ZS90 model). A series formulations, prepared with Capmul PG8, were found to exhibit relatively smaller globule size, lower PDI and higher ZP, in comparison to that of B series formulations, prepared with Capmul MCM (Table III). In A series, the formulations resulted in variable globules, with sizes from 145.3 ± 2.03 to 292.7.6 ± 2.68 nm, the PDI from 0.297 ± 0.02 to 0.461 ± 0.03 and ZPs from − 9.64 ± 1.22 to − 7.18 ± 0.99 mV. Similarly, in B series also, globule sizes were variable from 240.9 ± 4.74 to 288.9 ± 4.05 nm, while PDI ranged from 0.361 ± 0.05 to 0.501 ± 0.02 and the ZPs varied from − 11.7 ± 1.28 to − 10.68 ± 0.90 mV. These results indicated that, upon increasing the content of oil in the formulation, the droplet size was increased, which in turn reduced the surface area. Further, this might be due to the dependency on the surfactant concentration, i.e., increased oil content required increased quantity of surfactant to maintain the lower droplet size. Among all formulations of both series, the A1 formulation showed the least globule size, least PDI and not higher ZP. Hence, this was further studied.
Preparation and Characterization of Solid SMEDDS and Liquisolid Formulations
Preparation of Solid SMEDDS
Among liquid SMEDDS, A1 containing 15% of Capmul PG8 oil was selected for converting into solid SMEDDS, by adsorbing on different solid carriers, namely, Neusilin US2, Syloid XDP, Syloid 244 FP and Fujicalin SG. The developed solid SMEDDS were coded as SN, SX, SP and SF, respectively (Table IV).
Preparation of Liquisolid Formulations
From the drug solubility studies in different cosurfactant vehicles (Table I), the Transcutol P showed maximum Lov solubility. Four carriers like Neusilin US2, Syloid XDP, Avicel PH 102 and Fujicalin SG were utilized for conversion of liquid formulation into solid form and were coded as LN, LS, LA and LF, respectively. Aerosil 200 was included as coating material. The ratio of carrier and coating material was maintained at 10:1.
Characterization of Solid SMEDDS and Liquisolid Compacts
Determination of Liquid Retention Potential, Liquid Load Factor and Powder Flow Properties
Liquid retention potential was determined for all the four prepared solid SMEDDS. The liquid retention potential values of SN, SX, SP and SF were found to be 1.60, 1.60, 1.33 and 0.50, respectively (Table IV). Neusilin US2 and Syloid XDP showed similar liquid retention potential values. Based on this observation, either SN or SX solid SMEDDS could be considered as potential solid SMEDDS. Liquid load factor of liquisoid formulations, i.e., LS, LN, LA and LF were 1.64, 1.64, 0.51 and 0.51, respectively (Table V). Liquisolid formulations prepared with Syloid XDP (LS) and Neusilin US2 (LN) showed increased and similar liquid load factors when compared to that of LA and LF. Further, powder flow properties like angle of repose, Carr’s index and Hausner’s ratio were determined for both LS and LN. Due to the lower liquid load factor, and the requirement of more quantity of the carrier, the powder flow studies were not investigated for LA and LF. For all the developed solid SMEDDS, powder characteristics were found. SX, solid SMEDDS prepared with Syloid XDP, showed the least angle of repose among all the four prepared solid SMEDDS. SX solid SMEDDS exhibited the flow characters like angle of repose, Hausner’s ratio, Carr’s index and % assay values of 26.07 ± 0.35, 1.23 ± 0.04, 18.53 ± 1.81 and 98.58 ± 0.38%, respectively (Tables IV and VI). The lower angle of repose indicated the good flow behavior of the powder formulation. Therefore, solid SMEDDS developed with Syloid XDP was optimized based on flow characteristics. Among liquisolid formulations, the selected LS and LN formulations showed angle of repose, Hausner’s ratio and Carr’s index of 25.23 ± 0.78, 1.23 ± 0.02, 19.42 ± 1.65 and 38.33 ± 0.67, 1.34 ± 0.01, 25.61 ± 0.59, respectively (Table VII). Further, both were tested for in vitro dissolution also (Fig. 2). Among these two, LN was having more angle of repose, an indicator of poor flow. Hence, LS was assumed as optimized and used for further studies.

A comparison of the dissolution profiles of pure drug, solid SMEDDS (SX) and liquisolid formulations (LS and LN)
In vitro Dissolution Studies of Optimized Solid SMEDDS and Liquisolid Formulations
USP dissolution type II apparatus was used in this study. The dissolution test was done on optimized solid SMEDDS (SX), liquisolid formulations (LS and LN) and pure drug. All the formulations contained 10 mg of lovastatin. Among the SX, LS and LN formulations, there was no significant change at 120 min. But, significant difference was observed at 30 min. Here, pure drug showed 17.23 ± 0.58% at 30 min and 57.78 ± 0.57% release at 120 min. Here, LN, LS and SX showed 72.44 ± 1.40%, 93.80 ± 0.75% and 79.13 ± 0.95% release respectively at 30-min time point (Fig. 2). It clearly indicated the promising role of solid SMEDDS and liquisolid formulations for improving dissolution of Lov. Further, the solid SMEDDS and liquisolids showed significant improvements in the dissolution of Lov when compared. Solid SMEDDS (SX) was not faster when compared to liquisolid LS. Probably, particle wetting and surface areas were not same for both SX and LS formulations. After 45 min, both LS and SX behaved similarly. This was possible due to the transformation of insoluble crystalline form to more soluble form of drug during the formulation development, coupled with increased surface area of the generated solid particles, in both liquisolids and solid SMEDDS. Among the two liquisolid formulations, LS showed faster dissolution profile and also increased release at 15 min time point when compared to LN. Based on flow behavior and faster release, LS was selected for further study. In related previous studies on this drug (Lov), liquisolid tablets (25) showed below 40% drug release in the first 30 min. This variation could be due to the different excipients used. While, solid SMEDDS of Lov (27) at 15 min resulted around 50% drug release in another study. In comparison to previously reported and designed liquisolid compacts and solid SMEDDS, the optimal LS and solid SMEDDS of this study appeared to be somewhat better in in vitro release behavior.
Studies on Solid-State Characteristics
Differential Scanning Calorimetry
DSC is a thermal analytical method to monitor both endothermic (melting, phase transition, and chemical degradation) and exothermic processes. It is also used in the detection of drug- carrier interactions. Approximately 10 mg of the drug or formulation sample was employed here. The study was conducted in the temperature range of 20–250 °C at a heating rate of 10 °C/min to obtain thermograms of lovastatin pure drug, liquisolid formulation (LS) and solid SMEDDS (SX) (Fig. 3). Lov drug showed a sharp peak (endothermic) at 172.05 °C indicating its purity and existence in crystalline form. The thermograms of LS and SX did not show any kind of endothermic peaks, indicating that this drug substance was in dissolved/solution state in the developed formulations. Hence, specific peaks were missing in comparison to pure drug. Further, this could be due to the transformation of crystalline form into amorphous form in the final formulations or increased bulkiness by the added excipients.

An overlay of DSC thermograms. A Lovastatin drug. B SX (optimized solid SMEDDS). C LS (optimized liquisolid formulation)
Scanning Electron Microscopy
By SEM, the surface morphology was found for Lov drug, carrier Syloid XDP, and the formulations SX and LS. In SEM pictures (Fig. 4), drug lovastatin appeared as rod-like crystals. In both SX and LS formulations, rod-shaped crystals were not observed and it indicated the adsorption of dissolved drug (loss of crystalline nature) on the carrier, during formulation development.

SEM pictures of A lovastatin drug (× 100), B carrier Syloid XDP (× 60), C liquisolid formulation (LS) (× 250) and D solid SMEDDS (SX) (× 100)
Powder X-Ray Diffractometry of the Formulations
Powder X-ray diffraction spectra were obtained for drug, syloid XDP carrier and both optimal formulations SX and LS. Their overlaid spectra are presented in Fig. 5. The Lov drug showed 31 peaks and indicated the crystalline nature of drug. Among these, 3rd, 4th and 5th peaks were stronger and observed at 2-theta values (deg) of 16, 17 and 18, respectively. No similar peaks were detected in SX and LS formulations, which revealed that the drug had lost the crystallinity in both the developed delivery systems during formulation stage. The change in crystallinity of drug would influence the drug release from formulations. The obtained PXRD data is in good agreement with the DSC results. This behavior (loss of crystallinity) is expected to improve the solubility of the drug in GI fluids, resulting in a better bioavailability.

An overlay of PXRD spectra of A lovastatin drug, B carrier Syloid XDP, C liquisolid formulation (LS) and D solid SMEDDS (SX)
Stability Studies During Storage for Short Period
The prepared LS and SX solid formulations were stored for 90 days. The SX samples were checked periodically at monthly interval for globule size, PDI and ZP. Further, the drug content in both SX and LS formulations was assayed (Table VIII). In general, the SX formulation characters remained almost same with insignificant minor changes in size, ZP, PDI and drug content. Further, the content of LS was also not changed. Hence, both solid formulations were considered stable for 90-day period.
Pharmacokinetic Study and Data Analysis
Oral bioavailability (BA) study was conducted in male Wistar rats (n = 6) for coarse suspension, optimized formulations SX and LS. Limit of detection and quantification of Lov were 100 ng/ml and 250 ng/ml, respectively. The linearity was noticed within the concentration range of 0.25–16 µg/ml. The R2 was 0.997 value, for standard graph. The retention times of Lov and that of internal standard (IS) were 6.9 and 4.1 min, respectively. Pharmacokinetic analysis and its parameters were estimated with Kinetica 2000 software. Mean serum concentrations of Lov in serum vs time profile of all groups are given in Fig. 6. PK parameters such as AUC, Cmax, Tmax, t1/2 and MRT were calculated and expressed as mean ± SD (Table IX). One-way ANOVA was conducted with a software (version 5.03, Graph pad prism) for the statistical comparison. The SX exhibited improved PK values for the parameters of AUC, Cmax, t1/2 and MRT. These were also statistically significant, when compared with that of LS and CS. The bioavailability improvements of SX and LS were 2.57 and 1.43 fold, respectively, in comparison to that of CS. Further, SX was having 1.79 fold enhancement in oral BA when compared to that of LS. The increased bioavailability of LS and SX, when compared to CS was due to increased dissolution of drug. Here, the increased bioavailability of SX was due to the combined effect of faster drug release and absorption via portal circulation, as well as lymphatic transport. Whereas LS was not able to provide lymphatic transport, but contributed for faster drug release along with first-pass metabolism. This observation confirmed the previous observations made on solid SMEDDS vs liquisolids of iloperidone drug, i.e., a comparative study of liquisolid and solid SMEDDS of iloperidone drug revealed the superiority of solid SMEDDS over liquisolid (29). In addition, enhanced t1/2 and MRT values provided an evidence for the probable sustained effect/action of the designed solid SMEDDS (SX) when compared to LS. In a previous bioavailability study (50), a marketed tablet formulation (Cholilysis® 10 mg) was compared to a prepared nanosuspension, and in this study, no significant difference was noticed in AUCs of both.

Pharmacokinetic profiles—a comparison of lovastatin CS (coarse suspension), SX (solid SMEDDS) and LS (liquisolid) formulations
Study of Pharmacodynamic (Anti-hyperlipidemic) Activity
Triton X-100, a nonionic surfactant, was used to induce hyperlipidemia in Wistar rats as per reported method by injecting through i.p route (48). Triton-X-100 inhibits peripheral lipoprotein lipases, which remove the lipid particles from body. The observed lipid levels of all five groups of animals are given in Table X and Fig. 7. Here, normal control rats (group A) exhibited no changes in lipid levels throughout study period. But, in Triton X-100-treated control rats (group B), all lipid levels, namely, CHL (cholesterol), TG (triglycerides), LDL (low density lipoproteins) and VLDL (very low density lipoproteins) except HDL (high density lipoproteins) were found to increase after 24 h post-dose, and the increased lipid levels were sustained for a further period of 72 h and reached to normal levels after 72 h (see Table X). The mechanism of action of Lov is by inhibiting the HMG-CoA reductase, a key enzyme, responsible for the conversion of HMG-CoA to mevalonate in biosynthesis of cholesterol. The CS treatment showed a phase of decline in CHL, TG, LDL and VLDL levels after 4 h, and a plateau phase was noticed from 8 to 12th hour. Thereafter, again the lipid levels were increased after 24 h and sustained up to 48 h. This could be due to the loss of drug activity by metabolism. After 72 h of CS dosing, the lipid levels reached to that of control level, due to loss of Triton X-100 activity. LS treatment also showed a phase of decline in CHL, TG, LDL and VLDL levels after 4 h and a plateau phase was noticed from 6 to 12th hour. Thereafter, again the lipid levels increased after 24 h due to the lack of drug activity by metabolism. After 72 h post-dose of LS, the lipid levels reached to that of control level due to loss of Triton X-100 activity. Biphasic lipid profile was noticed for both CS and LS. In addition, the SX treatment showed a phase of decline in CHL, TG, LDL and VLDL levels at 2 h (i.e., early onset of anti-hyperlipidemic effect) and a plateau phase was noticed from 6th hour to 24 h, and further, SX showed relatively better performance over CS and LS in maintaining the reduced lipid levels for a prolonged period, i.e., up to 24 h. But, among CS and LS, the LS performance was slightly better in its anti-hyperlipidemic activity. In contrast, on HDL levels, the effect of LS, CS and SX was not significant and only contributed for slight increment in HDL levels when compared to that of hyperlipidemic control rats (group B). Among, CS, LS and SX, the SX showed early action and was lasting for at least 12 h longer period. Taken together, SX was considered as superior delivery system over that of LS in its action. In a previous comparative study (29), iloperidone solid SMEDDS and liquisolid formulations behaved similarly without change in PD effects, although an improvement in BA was observed for solid SMEDDS over that of liquisolid formulation. Whereas in another previous comparative study, fluvastatin solid SMEDDS (30) showed improved BA and elicited early action with longer duration over that of liquisolid formulation.

Comparative lipid profiles of normal control, hyperlipidemic control, lovastatin CS (coarse suspension), SX (solid SMEDDS) and LS (liquisolid formulation)
CONCLUSION
Lov is a low-dose BCS class II drug. It was reported to have 5% oral BA due to poor solubility and first-pass effect. To improve the oral BA of Lov, dissolution enhancement approaches like solid SMEDDS and liquisolid technique were tried. Lov solid formulations of SMEDDS and liquisolid were developed and characterized. DSC, PXRD and SEM studies confirmed the loss of crystallinity of drug during formulation development. The optimal SMEDDS (SX) and liquisolid compacts (LS) were found to have angles of repose of 26.07 ± 0.35 and 25.23 ± 0.78, respectively, an indication of excellent flow behavior. During in vitro dissolution studies, the LS showed faster dissolution when compared to solid SMEDDS at 15-min time point. Pharmacokinetic study on Wistar rats showed that there was 2.57 fold and 1.43 fold improvements in oral BA of solid SMEDDS and liquisolid formulations respectively in comparison to that of a coarse suspension. In PD study, Lov solid SMEDDS showed improved anti-hyperlipidemic efficacy when compared to liquisolid formulation in terms of early action and sustained effects for prolonged period. In future, the solid SMEDDS of Lov may be developed as marketed product after comparing the relative performance with currently marketed tablets. Taken together in this study, optimal solid SMEDDS and liquisolids of lovastatin were designed and developed. Further, their PK and PD studies revealed the superiority of solid SMEDDS of lovastatin.
References
Yingpeng T, Qin Z, Wen S, Jianxin W. Mechanisms of oral absorption improvement for insoluble drugs by the combination of phospholipid complex and SNEDDS. Drug Deliv. 2019;26:1155–66.
Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44:235–49.
Anuj GA, Ashok K, Paraag SG. Self emulsifying drug delivery system for enhanced solubility and dissolution of glipizide. Colloids Surf B. 2015;126:553–60.
Yanbin H, Wei-Guo D. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B. 2014;4:18–25.
Khames A. Liquisolid technique: a promising alternative to conventional coating for improvement of drug photostability in solid dosage forms. Expert Opin Drug Deliv. 2013;10:1–9.
Zhi HL, Asim KS, Paul WSH. Overview of milling techniques for improving the solubility of poorly water-soluble drugs. Asian J Pharm Sci. 2015;10:255–74.
Jens-Uwe AHJ, Müller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomed. 2008;3:295–309.
Mu H, René H, Müllertz A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int J Pharm. 2013;453:215–24.
Kazi M. Design of lipid-based formulations for oral administration of poorly water-soluble drug fenofibrate: effects of digestion. AAPS Pharm Sci Tech. 2012;13:637–46.
Craig DQM, Barker SA, Banning D, Booth SW. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int J Pharm. 1995;114:103–10.
Khaled A, Ayat AA, Mahmoud EB, Ahmed ME. Role of self-emulsifying drug delivery systems in optimizing the oral delivery of hydrophilic macromolecules and reducing inter individual variability. Colloids Surf B. 2018;167:82–92.
Caitriona MOD. Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci. 2002;15:405–15.
Fang L, Rongfeng H, Bin W, Yun G, Gang C, Song G, et al. Self-microemulsifying drug delivery system for improving the bioavailability of huperzine A by lymphatic uptake. Acta Pharm Sin B. 2017;7:353–60.
Dahan A, Amnon A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release. 2008;129:1–10.
Venkateswarlu K, Preethi JK, Chandrasekhar KB. Enhancement of loperamide dissolution rate by liquisolid compact technique. Adv Pharm Bull. 2016;6:385–90.
Fahim JS, Sachin LT, Umesh BK. Design and development of liquisolid compact of candesartan cilexetil to enhance dissolution. J Pharm Res. 2013;7:381–8.
Yadav VB, Yadav AV. Improvement of solubility and dissolution of indomethacin by liquisolid and compaction granulation technique. J Pharm Sci Res. 2009;1:44–51.
Anna C, Marta S, Aleksandra A, Emilia S, Katarzyna W. Development and evaluation of liquid and solid self-emulsifying drug delivery systems for atorvastatin. Molecules. 2015;20:21010–22.
Wu CY, Leslie ZB. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22:11–22.
Wang RW, Prasad HK, Anthony YHL, Paul ET, Peter GF, Kamlesh PV. Biotransformation of lovastatin: identification of cytochrome P450 3A proteins as the major enzymes responsible for the oxidative metabolism of lovastatin in rat and human liver microsomes. Arch Biochem Biophys. 1991;290:355–61.
Li J, Liuying D, Xu C, Weiwen J, Hongyu P, Gang C, et al. The characteristics and mechanism of coadministration of lovastatin solid dispersion with kaempferol to increase oral bioavailability. Xenobiotica. 2019;50:593–601.
Jun Z, Daxin Z. Improvement of oral bioavailability of lovastatin by using nanostructured lipid carriers. Drug Des Dev Ther. 2015;9:5269–75.
Suresh G, Manjunath K, Venkateswarlu V, Satyanarayana V. Preparation, characterization, and in vitro and in vivo evaluation of lovastatin solid lipid nanoparticles. AAPS Pharm Sci Tech. 2007;8:E1–9.
Manjil P, Avinash T, Surendra G, Sanjay S. Solubility enhancement of lovastatin by modified locust bean gum using solid dispersion techniques. AAPS Pharm Sci Tech. 2008;9:1262–9.
Lakshmi P, Abbulu K. Formulation and evaluation of lovastatin tablets by using liquid solid compact technique. GSC Biological and Pharmaceutical Sciences. 2019;8:139–55.
Maulik JP, Natvarlal MP, Ritesh BP, Rakesh PP. Formulation and evaluation of self-microemulsifying drug delivery system of lovastatin. Asian J Pharm Sci. 2010; 5: 266–75.
Qureshi MJ, Chitneni M, Wong GK. Enhancement of solubility and therapeutic potential of poorly soluble lovastatin by SMEDDS formulation adsorbed on directly compressed spray dried magnesium aluminometasilicate liquid loadable tablets: a study in diet induced hyperlipidemic rabbits. Asian J Pharm Sci. 2015;10:40–56.
Suparna SB, Jasmine GA. Development and evaluation of liquid and solid self-microemulsifying drug delivery system of lovastatin. Asian J Pharm. 2016;10:22–35.
Dinesh S, Arjun N, Kishan V. Development, characterization, comparative pharmacokinetic and pharmacodynamic studies of iloperidone solid SMEDDS and liquisolid compact. Drug Dev Ind Pharm. 2020;46:587–96.
Madhav KV, Kishan V. Improvement of anti-hyperlipidemic activity and oral bioavailability of fluvastatin via solid self-microemulsifying systems and comparative with liquisolid formulation. Curr Drug Deliv. 2017;15:1245–60.
Madhav KV, Kishan V. Self microemulsifying particles of loratadine for improved oral bioavailability: preparation, characterization and in vivo evaluation. J Pharm Investig. 2017;48:497–508.
Ramesh J, Muzammil AS, Prabhakar K, Kishan V. Development of a self-microemulsifying drug delivery system of domperidone: in vitro and in vivo characterization. Acta Pharm. 2013;63:241–51.
Hamidi M, Najmeh Z, Mohammad AS. A simple and sensitive hplc-uv method for quantitation of lovastatin in human plasma: application to a bioequivalence study. Biol Pharm Bull. 2009;32:1600–3.
Prapaporn B, Karen K, Anja G, Thomas R, Varaporn BJ. Characterization of microemulsion structures in the pseudoternary phase diagram of isopropyl palmitate/water/Brij 97:1-butanol. AAPS Pharm Sci Tech. 2006;7:E1–6.
Sourav B. DLS and zeta potential – what they are and what they are not? J Cont Release. 2016;235:337–51.
Venu MK, Kishan V. Cationic solid self micro emulsifying drug delivery system (SSMED) of losartan: formulation development, characterization and in vivo evaluation. J Drug Deliv Sci Tech. 2016;35:190–9.
Spireas S, Tao W, Rakesh G. Effect of powder substrate on the dissolution properties of methyclothiazide liquisolid compacts. Drug Dev Ind Pharm. 1999;25:163–8.
Fahmy RH, Mohammed AK. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In vitro and in vivo evaluation. Eur J Pharm Biopharm. 2008;69:993–1003.
Ileleji KE, Zhou B. The angle of repose of bulk corn stover particles. Powder Technol. 2008;187:110–8.
Rakhi BS, Mobin AT, Mansoor AK. Comparative evaluation of flow for pharmaceutical powders and granules. AAPS Pharm Sci Tech. 2008;9:250–8.
Pooria G, Tahereh TM, Bijan R. Differential scanning calorimetry techniques: applications in biology and nanoscience. J Biomol Tech. 2010;21:167–93.
Cooper A. Microcalorimetry and related techniques. Edited by Robert S. Marks, David C. Cullen, Isao Karube, Christopher R. Lowe and Howard H. Weetall. John Wiley & Sons. 2007.
Andreza R, Ana F, Delfim S, Francisco V. Preparation and solid-state characterization of inclusion complexes formed between miconazole and methyl-β-cyclodextrin. AAPS Pharm Sci Tech. 2008;9:1102–9.
Vinay Kumar V, Chandrasekar D, Sistla R, Kishan V, Yamsani MR, Prakash VD. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: Influence of wax and glyceride lipids on plasma pharmacokinetics. Int J Pharm. 2007;335:167–75.
Scheubel E, Lindenberg M, Beyssac E, Cardot JM. Small volume dissolution testing as a powerful method during pharmaceutical development. Pharmaceutics. 2010;2:351–63.
Gibaldi M, Stuart F. Establishment of sink conditions in dissolution rate determinations: theoretical considerations and application to non disintegrating dosage forms. J Pharm Sci. 1967;56:1238–42.
Nahas K, Provost JP. Blood sampling in the rat: current practices and limitations. Comp Clin Path. 2002;11:14–37.
Adigun NS, Oladiji AT, Ajiboye TO. Antioxidant and anti-hyperlipidemic activity of hydroethanolic seed extract of Aframomum melegueta K. Schum in Triton X-100 induced hyperlipidemic rats. S Afr J Bot. 2016; 105: 324–32.
Narendar D, Kishan V. Improved anti-hyperlipidemic activity of Rosuvastatin Calcium via lipid nanoparticles: pharmacokinetic and pharmacodynamic evaluation. Eur J Pharm Biopharm. 2017;110:47–57.
Dalia AG. Nanoparticles of lovastatin: design, optimization and in vivo evaluation. Int J Nanomedicine. 2020;15:4225–36.
Funding
The authors express thanks to AICTE (All India Council for Technical Education), Govt. of India, New Delhi, for sanctioning financial assistance under Research Promotion Scheme, vide F.No. 8–77/RIFD/RPS (POLICY-1)/2016–17, to carry out this research study.
Author information
Authors and Affiliations
Contributions
Both the authors have equally contributed for this research work. The research idea was proposed by Prof. V. Kishan (corresponding author). The experimental work was carried out by Dinesh Suram, a Ph.D scholar (first author). Prof. V. Kishan interpreted the data and supervised the study.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Suram, D., Veerabrahma, K. Design and Development of Solid SMEDDS and Liquisolid Formulations of Lovastatin, for Improved Drug Dissolution and In vivo Effects—a Pharmacokinetic and Pharmacodynamic Assessment. AAPS PharmSciTech 23, 123 (2022). https://doi.org/10.1208/s12249-022-02272-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-022-02272-2