
Abstract
The current research work aims to study the pharmacokinetic and nasal ciliotoxicity of donepezil liposome–based in situ gel to treat Alzheimer’s disease. The physicochemical properties and first-pass metabolism of donepezil HCl result in low concentrations reaching the brain post oral administration. To overcome this problem, donepezil HCl–loaded liposomes were formulated using the ethanol injection method. The donepezil HCl–loaded liposomes were spherical with a size of 103 ± 6.2 nm, polydispersity index of 0.108 ± 0.008, and entrapment efficiency of 93 ± 5.33 %. The optimized in situ gel with donepezil HCl–loaded liposomes showed 80.11 ± 7.77 % drug permeation than donepezil HCl solution–based in situ gel (13.12 ± 4.84 %) across sheep nasal mucosa. The nasal ciliotoxicity study indicated the safety of developed formulation for administration via nasal route. The pharmacokinetics and biodistribution study of developed formulation showed higher drug concentration (1239.61 ± 123.60 pg/g) in the brain after nasal administration indicating its better potential via the nasal pathway. To treat Alzheimer’s disease, the administration of liposome-based in situ gel through the nasal pathway can therefore be considered as an effective and promising mode of drug delivery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Alzheimer’s disease (AD) has become a severe public health issue, especially in geriatric population. Alzheimer’s disease, which accounts for an estimated 60–80% of dementia cases worldwide, is the leading cause of dementia (1). Alzheimer’s disease cannot be cured, and current treatments focus on decreasing the rate of disease progression besides managing the symptoms (2). In addition to the unavailability of drugs to cure the disease, delivering existing drug molecules in the brain is also a major challenge (3). Various strategies are being studied for the delivery of neurotherapeutic agents across the blood-brain barrier. However, more efforts are required to accomplish this goal (4,5,6). The present work addresses the challenges of delivering donepezil HCl safely and effectively into the brain.
Donepezil HCl is a specific and reversible acetylcholinesterase inhibitor. The anti-cholinesterase enzyme is responsible for the breakdown of acetylcholine. The concentration of acetylcholine in the brain is increased by donepezil HCl, which results in enhanced cholinergic activity. Acetylcholine is associated with memory and learning, and it is deficient in patients with Alzheimer’s disease.
The downside of current oral treatment options comprise high frequency of administration, first-pass metabolism, and side effects associated with gastrointestinal tracts like nausea, diarrhea, anorexia, muscle convulsion, and low concentration of drug reaching the brain (7). Researchers have developed lipid-based nanoformulation delivering a drug to the brain, like liposomes, solid lipid nanoparticles, nanostructured lipid carriers, nanoemulsions, niosomes, and cubosomes via nasal route. They reported a significant role of lipidic formulation in the improvement of the drug delivery via nasal route. This formulation is effective in protecting the drug from chemical and biological breakdown and P-gp efflux proteins responsible for reduced bioavailability (8, 9).
We attempted in this study to use liposomes as a carrier for donepezil HCl and evaluated nasal route of administration to overcome first-pass metabolism, along with GI side effects associated with the conventional oral route to keep plasma concentration in therapeutic range (10). Liposomes may target the brain by employing various transport molecules present on the surface of brain capillary endothelial cells (BCECs) (11). The liposomal formulation components and their structure play a significant role in improving pharmacokinetics and bioavailability. Lipids like cholesterol and phosphatidylcholine have the exact nature as biological membranes (12). This facilitates the permeation of liposomes through the cell membrane.
It is theorized that intranasal administration has a higher utility in delivering therapeutic agents to the CNS than routes requiring crossing the BBB for various reasons, as reported in earlier literature (13, 14). The intranasal administration has potential advantages, such as patient self-administration thereby improving patient compliance, rapid initiation of action, minimizing systemic exposure, and decreasing peripheral adverse effects. Additionally, it also enhances drug bioavailability in the brain. The drug is partly administered to the brain through the olfactory or trigeminal route after nasal administration of the formulation, thereby preventing first-pass metabolism (15). The olfactory and trigeminal nerve served as a direct pathway for the permeation of drugs from the nasal cavity region to the brain. The drug’s passage via olfactory neurons occurs by passive diffusion for small lipophilic compounds and paracellular or endocytosis for large and hydrophilic molecules (16, 17).
Conversely, the trigeminal pathway develops a connection between the nasal cavity and cerebrum with the pons region of the brain and frontal cortex (16, 18). The limited surface area of the nasal cavity can be a challenge; however, the drug absorption can be facilitated by the existence of high vascularization (19). Absorption of the drug can further be enhanced by increasing the formulation’s site retention time by in situ gelation.
The present research aimed to develop and characterize stable donepezil HCl–loaded liposomes with a vesicular size of around 100 nm to facilitate absorption after intranasal administration. The developed liposomes were incorporated into a formulation that forms an in situ gel, allowing easy administration while ensuring longer retention at the olfactory site in the nasal cavity. The selected optimized formulation was evaluated for in vivo performance using an animal model, and its performance was compared with the marketed product approved for oral administration.
MATERIALS AND METHODS
Materials
Donepezil HCl was obtained as a gift sample from the Ranbaxy laboratory (Gurgaon, India). Hydrogenated soy phosphatidyl choline (HSPC) and cholesterol were donated by Lipoid (Newark, USA). Sucrose and ammonium sulfate were purchased from Merck (Mumbai, India). Histidine and ethanol were purchased from Merck (Mumbai, India). Gellan gum and xanthan gum were received as a gift sample from C. P. Kelco (Mumbai, India). All analytical reagent grade chemicals and reagents were used for the study. All chromatographic grade solvents were used for analysis.
Methods
Preparation of Donepezil HCl–Loaded Liposomes
Ammonium sulfate gradient–based active loading technique similar to the one used for encapsulating doxorubicin HCl into liposomes (Doxil) (20) was used for encapsulating donepezil HCl into liposomes. An accurately weighed quantity of hydrogenated soy phosphatidylcholine (HSPC) and cholesterol were dissolved in ethanol. An aqueous solution of ammonium sulfate solution was prepared. The ethanolic lipid solution was injected into preheated ammonium sulfate solution at 60–65°C through a needle (26 μm diameter) with continuous stirring. To obtain blank multilamellar vesicles (MLV) and extract the solvent, the solution was stirred for 1 h at 60–65°C. Following this, multilamellar vesicles (MLV) were passed through the high-pressure homogenizer (HPH) (Panda plus, Niro sovai, Japan, Flow rate: 3.7 ml/s; type of valve: S shape (flat shape); material of construction (MOC) of the valve is ceramic)) to obtain the large unilamellar vesicles (LUV) having size around 100 nm. During the HPH cycle, the cold water was circulated continuously to prevent product temperature rise.
Sucrose solution (10%) was used to remove the unencapsulated ammonium chloride from the blank liposomes to produce an ammonium sulfate gradient. In brief, 10 ml of blank liposome suspension was added in a dialysis bag [molecular weight cut off (MWCO) 10 kDa, Himedia Limited, India, and placed in a 2000-ml beaker consisting of 10% sucrose solution. The magnetic stirrer was used at a speed of 1000 rpm and an interval of 1 h; sucrose solution was removed, checked for conductivity, and replaced with fresh solution. This process is continued for 24 h until conductivity reached below 10 μS/cm. The conductivity below 10 μS/cm indicated the complete removal of free ammonia from blank liposomes. After that, the donepezil HCl solution was added to the blank liposomes and stirred for 1 h at 65°C to enable drug loading. The formed liposomes were cooled in an ice bath for 15 min. Finally, a liposome dispersion was stored at 2–8°C till further analysis (21, 22). The composition of liposomal batches (batch size-100 ml) is presented in Table I.
HPLC Analytical Method
The samples were quantified by injecting them into the Merck licosphere LiChrosper C18 RP end–capped column (250 mm × 4.6 mm, internal diameter 5 μm) maintained at 25°C. The mobile phase is composed of a mixture of methanol: phosphate buffer (pH 3.5 adjusted with 0.5 % v/v orthophosphoric acid solution in distilled water) (60:40 % v/v) with flow rate 1 ml/min and detected at 230 nm wavelength (23).
Characterization of Liposomes
Vesicle Size and Polydispersity Index
The average hydrodynamic vesicle size and polydispersity index of liposomes were determined using Malvern Zetasizer (NANO ZS 90, Malvern Instruments Ltd., Malvern, UK). Samples were used without any dilution. All the parameters were measured in triplicate at 25 ± 2°C at an angle of 90° (24).
Zeta Potential
The zeta potential of the developed donepezil HCl–loaded liposomes was determined in pre-rinsed standard special shape disposable cuvettes equipped with electrodes. Zeta potential was determined by photon correlation spectroscopy and is noted as the Z-average. All measurements were carried out in triplicates (25, 26).
Determination of Drug Loading
The percentage of donepezil HCl entrapped in the liposome was determined by centrifugation using Amicon® Ultra filter centrifugal tubes (30 KDa, 4 ml capacity) (Merck, India). Briefly, 1 ml of donepezil HCl–loaded liposomal suspension was centrifuged at 10,000 rpm for 10 min at 25°C using a vacuum pump to separate the drug entrapped in liposomes from the free drug. The eluted fraction was collected from the bottom of the device. The washings were conducted using histidine buffer pH 6.5 repeatedly to collect free drugs. The free drug solution collected at the bottom of the device was appropriately diluted and filtered through a 0.22 μ filter and analyzed (24, 27,28,29). The drug loading was calculated using following Eq. (2)
Entrapment Efficiency
The percentage of donepezil HCl entrapped in the liposome was determined by centrifugation using Amicon® Ultra filter centrifugal tubes (30 KDa, 4 ml capacity) (Merck, India). Briefly, 1 ml of donepezil HCl–loaded liposomal suspension was centrifuged at 10,000 rpm for 10 min at 25°C using a vacuum pump to separate the drug entrapped in liposomes from the free drug. The eluted fraction was collected from the bottom of the device. The washings were conducted using histidine buffer pH 6.5 repeatedly to collect free drugs. The free drug solution collected at the bottom of the device was appropriately diluted and filtered through a 0.22 μ filter and analyzed (24). Entrapment efficiency was calculated by Eq. (1)
Morphological Characterization of Liposomes
Cryo High-Resolution Transmission Electron Microscopy
To study the size of the liposomes, cryo TEM analysis of the optimized batch (DL 5) was done using a negative staining method by transmission electron microscope (JEOL, JEM 2100, HRTEM, Japan). Fifty microliters of the liposomal suspension was placed on a carbon-coated copper grid. And the extra solution was removed with the help of filter paper. It was stained using 1% (w/v) aqueous phosphotungstic acid solution and dried at room temperature. Finally, the sample was placed in the transmission electron microscope, and micrographs were observed at 100 kV (30).
Preparation of Liposome-Based In Situ Gel
The liposome-based in situ gel was prepared using the “cold method”, explained by Pachis et al. (31). A clear and transparent gellan gum solution (0.5 % w/v) was obtained by heating at 90°C with constant stirring (mechanical stirrer, RQ-127A, India) cooled below 40°C. At the same time, the xanthan gum (0.15% w/v) solution was prepared with constant stirring using distilled water. Finally, drug-loaded liposomes were added to the xanthan gum solution, and the resulting mixture was added to the gellan gum solution and mixed for 15 min using a stirrer.
Characterization of In Situ Gel
pH
The pH of the developed in situ gel was determined using a digital pH meter (Hanna, HI, India). The average of three readings was recorded (n=3) (32).
Critical Ionic Concentration
The critical ion concentration was determined by combining 1 ml of the formulation with various simulated nasal fluid volumes (composition—7.45 mg/ml NaCl, 1.29 mg/ml KCl, and 0.32 mg/ml CaCl2.2H2O, pH-6.5) (33) in vials kept in a water bath at 32 ± 2°C. After a minute, vials were rotated and checked for the formation of a gel. The minimum volume of simulated nasal fluid required for gel formation was considered critical ionic concentration (34).
Gelling Time
The gelling time was determined by mixing 1 ml of in situ gel and a specific amount of simulated nasal fluid (pH 6.5), and the formation of the gel was visually observed. Finally, the time needed for in situ gel formation was noted (n=3) (35).
In vitro Mucoadhesion Testing
The prepared in situ gel’s mucoadhesive property was recorded using a texture analyzer (Brookfield, QTS, USA). The freshly isolated sheep nasal mucosa was stuck to the upper movable probe with cyanoacrylate glue. The in situ gel (1 ml) was kept onto the lower stationary stage with 0.3 ml of SNF. The probe holding the mucosa was lowered onto the in situ gel’s surface using a downward force of 5 g. The probe was kept in contact with in situ gel without any force for 60s to confirm enough contact between the nasal mucosa and in situ gel. Then at a constant speed of 30 mm/s, the probe was pushed vertically upwards, and the force needed to remove the nasal mucosa from the in situ gel was reported directly from the software. The test was conducted at 32 ± 2°C (36).
Viscosity
The viscosity of the formulated in situ gel before and after gelation was measured using a viscometer (Brookfield Engineering Labs, DV-I, USA). The 25-ml sample was taken in a glass tube. Spindles 18 and 96 were used at 10 rpm speed at 32 ± 2°C for viscosity determination before and after gelation, respectively. All readings were measured in triplicate (37).
In vitro Permeation Study
In situ gel’s in vitro permeation analysis was performed using sheep nasal tissue mounted on Franz diffusion cell (Orchid scientific limited, FDC-06, India). Sheep nasal mucosa was placed on a Franz diffusion cell with a permeation area of 4.52 cm2. Simulated nasal fluid (7 ml) (pH 6.5) was held in the receptor compartment. After conditioning for 20 min, 1 g of in situ gel was added to the donor segment of the diffusion cell. At 50 RPM, the entire system was maintained at 32 ± 2°C. A 0.5-ml sample was extracted from the receptor compartment at various time intervals and substituted with 0.5 ml of fresh simulated nasal fluid (pH 6.5). The amount (cumulative) of drug penetrated across the nasal tissue was calculated. The permeation profile was then compared with in situ gel containing donepezil suspension (30).
Stability Study
Stability analysis was conducted for batches DL4 and DL5. A suitable amount of liposomes in a closed high-density polyethylene bottle, was kept in the refrigerator (2–8°C). At different time intervals 0, 90, and 180 days, samples were characterized for particle size, drug loading, and entrapment efficiency (38).
Nasal Ciliotoxicity Studies
A nasal ciliotoxicity study was conducted using excised sheep nasal mucosa. For this purpose, fresh nasal mucosa was carefully excised from the nasal cavity of sheep at the local slaughterhouse with prior permission from concerned authorities of Ahmedabad Municipal Corporation Slaughter House, Jamalpur, Ahmedabad, stored in saline solution. The nasal mucosa was isolated from the septum and the connective tissue. Most aligned cartilaginous tissue was carefully separated with forceps and scissors without damaging or scratching the nasal mucosa. The isolated mucosa was preserved in phosphate buffer saline pH 6.4 during transport and used within 4 h (39). It was cleaned with a saline solution thrice. A portion of the nasal mucosa was exposed and treated with blank gel and liposome-loaded in situ gel. Phosphate buffer solution pH 6.4 and isopropyl alcohol (nasal mucociliary toxicity compound) were negative and positive control respectively. After 8 h of exposure, the nasal septum with the epithelial cell membrane was stained with hematoxylin for morphological examination. The sample was examined under an electron microscope at a resolution of 100× (Olympus, CX21FS1, Japan) (40).
Pharmacokinetic Study
Pharmacokinetic studies were performed on Sprague Dawley rats weighing 200 ± 50 g. The animal study protocol was approved by the Institutional Animal Ethical Committee (IP/PCEU/PHD/18/017). At 20 ± 2°C, the rats were kept on a 12-h light and dark period. All the rats were provided with standard laboratory feed. The rats fasted overnight before the beginning of the pharmacokinetic study.
As per the protocol, the rats were categorized into two groups. The rats of group I received 1 mg/kg body weight donepezil HCl liposome–based in situ gel by nasal route with micropipette with smooth tubing attached to it. To ensure proper retention of the formulation at the administration site, the rats were kept in an upright position for a few seconds (45–60 s) (41). On the other hand, group II animals were administered with marketed formulation (equivalent to 1 mg/kg body weight of donepezil HCl) using oral gavage up to the esophageal region via the oral route(42). Blood samples were collected from each of the six rats at 0.5, 1, 2, 4, 6, and 8 h. Post blood sample collection, all animals were sacrificed to collect vital organs such as the brain, lung, liver, heart, spleen, and kidneys for analysis of donepezil HCl. To separate plasma, blood was centrifuged, and all the organs were washed three times with saline, wiped, weighed, and stored at −80°C until further analysis. All organs were cut into small pieces and homogenized by adding 10% w/v phosphate buffer pH 7.4 with tissue homogenizer (Popular Trades, PT 194, Ahmedabad) (24).
The drug was extracted from plasma and tissue samples by an extraction method (liquid–liquid) to determine donepezil HCl. Exactly 100 μl of plasma or tissue homogenate was mixed with 100 μl of sodium chloride solution; vortexed (Remi, CM 101 plus, Mumbai, India) for 2 min and allowed to stand for 5 min. As a result, a mixture of 1 ml of methanol was added and mixed for 2 min. The mixture was centrifuged at 4000 rpm for 10 min at 4°C, and the supernatant was collected (43). The supernatant was filtered via a 0.45 μ filter. The amount of the drug was determined using the high-performance liquid chromatography (HPLC) (Jasco, LC-4000, Japan) with a UV detector.
The pharmacokinetic parameters were calculated by Kinetica software (non-compartment modeling) (Thermo Fisher Scientific, Waltham, USA, trial version 5.0), and statistical analysis was performed by GraphPad Prism statistical software. The time to reach maximum concentration (Tmax), maximum drug concentration (Cmax), area under the curve from time zero to 8 h (AUC 0–8), area under the curve from time zero to infinity (AUC 0–∞), area under the mean curve (AUMC 0–8), and elimination rate (Kel) were determined. The drug targeting efficiency (DTE %) was calculated using the following equation (44, 45),
where AUCbrain and AUCblood are the area under the brain tissue concentration-time and blood concentration-time curves, respectively.
Oxidative Stress Parameters
For the study, Sprague Dawley rats were selected and divided into four groups of six animals under normal control; group A, group B, and group C. They were injected with scopolamine (1mg/kg except in the normal control group) via an intraperitoneal route for 28 days to induce a condition similar to Alzheimer’s disease. After 28 days, animals in group A (scopolamine-induced amnesia group without any formulation); group B (in situ gel containing liposomes equivalent to 1 mg/kg body weight of donepezil HCl and administered by the intranasal route); group C (donepezil-marketed tablet equivalent to 1 mg/kg body weight of donepezil HCl and administered by oral route) were administered with formulations. The brains were collected from euthanized animals and washed in phosphate buffer pH 7.4 several times. Tissue homogenate was prepared with phosphate buffer pH 7.4 using a homogenizer (RQ 127, Remi Instruments, India) at 2500 rpm. The resulting mixture was centrifuged for 10 min at 4°C at 4000 RPM. The supernatant was collected, filtered through a filter of 0.45 μ, and used to determine different parameters such as acetylcholinesterase (AChE activity), sodium dismutase (SOD), and catalase activity.
Acetylcholinesterase (AChE) activity was determined by colorimetric analysis. The formation of the yellow anion of 5-thio-2-nitrobenzoic acid was determined, and the AChE level was calculated as a unit per microgram of protein. The modified Ellman technique was used to determine protein concentration using bovine serum albumin as a standard (46, 47).
Sodium dismutase (SOD) was determined by forming hydroxylamine nitrite by the oxidation of oxymine as per the standard method (48). Reduced glutathione (GSH) level was studied spectrophotometrically using hydrogen peroxide (49).
Catalase activity was estimated as described by Hugo et al. (50). In the boiling water bath, the malondialdehyde (MDA) was determined using thiobarbituric acid (0.67 % w/v). The resultant mixture’s pink color was analyzed, and the results were expressed as nmol/g protein (51).
Statistical Study
Statistical analysis was done using graph pad prism (GraphPad Software Inc., La Jolla, CA) software to compare all the data. All results were represented as mean ± SD. One-way ANOVA was used to calculate the significant difference between individual groups, followed by Tukey’s multiple comparison test. A p value of < 0.05 was considered statistically significant (52).
RESULTS AND DISCUSSION
Preparation and Characterization of Liposomes
The blank liposomes were prepared, and the effect of the ratio of HSPC to cholesterol was studied. It was observed that the HSPC to cholesterol ratio of 3:1 resulted in better liposomes. The probe sonication and high-pressure homogenization were evaluated for vesicle size reduction. The probe sonication for more than 2 min generated lots of heat and could not reduce the vesicle size to around 100 nm. Further, this process cannot be scaled up, so high-pressure homogenization (HPH) was selected. Many HPH cycles were required to reduce the vesicle size at 4–6°C temperature during preliminary trials. However, it can be seen in Table I that with an increase in temperature up to 60–65°C, the number of HPH cycles can be reduced to 20 (batch DL3). The liposomes were cooled using an ice bath immediately after size reduction.
The vesicle size is an important parameter related to physical stability and drug permeation across the biological membranes. Additionally, size is also vital to prevent uptake by the reticuloendothelial system (53). The polydispersity index (PDI) is a parameter to indicate size distribution, and thus, the uniformity of vesicles. It is considered a monodisperse formulation when the polydispersity index equals or less than 0.3 (54,55,56). The blank liposomes (batches DL1 to DL3) showed PDI in the range of 0.1 to 0.2, indicating uniformity of vesicles.
The charge present on the surface of the vesicles could impact the mechanism involved in their passage from the nasal cavity to the brain. The study reports that anionic nanoparticles follow the olfactory pathway and cationic follow the trigeminal pathway (57). It indicates the role of the surface charge of nanoparticles in defining the major drug transport pathway for brain targeting. The developed donepezil HCl liposomes bear a negative charge, and thus, it may follow the olfactory pathway to target brain for management of Alzheimer’s disease. The charge on liposomes is a function of various factors like ionic strength in the external environment of liposomes and type and concentration of phospholipid used. The zeta potential of formulated batches was near to −30 mV, which is enough to prevent agglomeration of such colloidal systems (58).
The active (remote loading) and passive loading methods are generally used to load drugs into the liposomes (59). The active loading method involves drug loading into the blank liposomes and mixing a liposome suspension with a drug solution, and drug encapsulation is driven by a transmembrane electrochemical gradient (59,60,61,62). This technique can result in high drug entrapment efficiency, less wastage of active compounds (< 5%) in manufacturing, and also increased stability during storage and administration (59, 63). Similar results were observed by Vikili-Ghartavol et al. for docetaxel liposomes prepared by remote loading technique. They reported enhanced drug delivery to the tumor and higher permeability and retention effect (EPR) (64).
Similarly, donepezil HCl was loaded by the ammonium sulfate gradient method in blank vesicles formed by high-pressure homogenization. The loading of donepezil HCl through a salt gradient effect was considered an effective and most advanced drug loading method with high encapsulation efficiency. USFDA has suggested this method for the formulation of Doxorubicin HCl liposomes {Doxorubicin, 2017 #1083}(20). This method differs from most other drug loading methods since liposome preparation neither required acidic pH nor alkaline pH (65). After size reduction, ammonia outside the liposomes was removed by repetitive dialysis using a 10% sucrose solution. Thus, a pH gradient is created. By creating a stable pH gradient, donepezil HCl can be protonated in the liposomes’ acidic interior. The proton pool is formed, which acts as a driving force for further drug loading. Thus, donepezil HCl loading is driven by protonation and charging of donepezil HCl within the liposomes (66). Further, the ionized drug inside the vesicle cannot diffuse from the lipidic bilayer, and drug leakage can be prevented. The drug loading time and pH were also studied and concluded that 1 h time and 5.5 pH were the optimum conditions for achieving higher encapsulation efficiency (data not shown).
The effect of ammonium sulfate concentration was also studied and found that 400mM concentration showed higher drug entrapment in the vesicles (batches DL4 and DL5). However, a further increase in the ammonium concentration did not show improvement in drug entrapment. Thus, a 400 mM concentration of ammonium sulfate was selected. We have evaluated dowex resin, hydrophilic-lipophilic balance (HLB) cartridge, ultracentrifugation, and Amicon® Ultra filter centrifugal devices for the same. Finally, the determination of free drug concentration was carried out using HPLC analysis after separating the free and encapsulated drug by Amicon® Ultra filter centrifugal devices (67). We could achieve 93% drug encapsulation after optimizing all the process and formulation parameters in batch DL 5. The ammonium sulfate gradient helps donepezil HCl to reach the inner core of the liposomes and achieve higher encapsulation. The protonation of the drug inside the liposomes prevent the leaching of the drug in the external environment.
Morphological Characterization of Liposomes
Cryo High-Resolution Transmission Electron Microscopy
To gather more information about the vesicle size and morphology cryo high-resolution transmission electron microscopy was carried out, and the result is shown in Fig. 1. The TEM micrograph showed that liposomes were spherical with uniform size distribution and no aggregation of vesicles. The unilamellar structure of vesicles is visible in the TEM image. The average vesicle size obtained from the TEM study was 97 nm (52).

Cryo HR TEM image of optimized batch (batch DL 5)
Formulation and Evaluation of the Liposome-Based In Situ Gel
The developed liposomes cannot be administered accurately in the nasal cavity. Further, the olfactory lobe’s retention time is crucial to achieving higher drug concentrations in the brain. After evaluating different gelling agents, the selected batches (DL4 and DL5) were formulated using gellan gum and xanthan gum as in situ gel. The batches were selected based on the number of homogenization cycles, particle size, and encapsulation efficiency. The selected batches (DL 4 and DL 5) required lesser homogenization cycles (18) so as to achieve desired particle size (around 100 nm) and higher encapsulation efficiency (> 85%).
The batches were prepared with varying concentrations of gellan gum solution (0.25%, 0.5%, and 1% w/v) and 0.15% w/v concentration of xanthan gum. The gel prepared using a lower gellan gum amount (0.25% w/v) was not enough to form a desirable gel. Furthermore, the highest gellan gum concentration (1% w/v) resulted in the hard gel. The degree of mucoadhesive strength depends on gellan gum solution concentration. The donepezil HCl–loaded liposomes were added into the 0.5% w/v gellan gum solution with a 0.15% w/v concentration of xanthan gum and then mixed with simulated nasal fluid the clear, transparent in situ gel with sufficient mucoadhesive strength.
The formulated in situ gel was evaluated for pH and was 5.0 ± 0.21 and 5.4 ± 0.32 for batches DL 4 and DL 5, respectively.
The nasal cavity contains approximately 0.1 ml of mucus, including sodium, potassium, and calcium ions (68). Therefore, it is imperative to estimate the quantity of simulated nasal fluid needed to form a gel. The best gel was considered the in situ gel, which needed a minimum amount of simulated nasal fluid for gelation. The results of both the formulations are shown in Table II. Batch DL5 showed 0.30 ± 0.05 % critical ionic concentration, indicating gel formation with a very low nasal fluid volume.
The batches were also studied for the time required for the formation of a gel post nasal administration. The time needed for the formation of gel must be significantly less to prevent loss of the formulation. The gelling time was 39 ± 4.33 s and 28 ± 2.85 s respectively for DL 4 and DL 5 batches, respectively.
To extend the drug release, the mucoadhesion is important property. It was determined to confirm the adhesion of the formulation to the nasal mucosa. The higher mucoadhesive strength was observed in liposome-based in situ gel due to interaction between formulation components and mucin of the nasal mucosa (40). The optimized batch adheres to the mucosa, and the mucoadhesive strength was 2320 ± 75 dyne/cm2, as shown in Table II.
The viscosity of the formulation also plays a vital role in its easy administration. The formulation must build up its viscosity after interaction with the simulated nasal fluid. In addition to that, the formed gel should preserve its integrity to facilitate the drug release for the desired period without dissolving or eroding. Gellan gum gels quickly in the presence of ions present in the simulated nasal fluid, and therefore, rapid in vivo gelation is expected (69). The viscosity of prepared formulations before gelation and after gelation is reported in Table II.
Figure 2 shows the nasal permeation profile of optimized in situ gel (batch DL 5) and Donepezil HCl solution–based gel. The permeation studies showed that the donepezil HCl–loaded liposome-based in situ gel revealed a higher (80.11 ± 7.77%) percentage drug permeation than donepezil HCl solution–based gel (13.12 ± 4.84%). In contrast to donepezil HCl solution–based gel, the optimized in situ gel showed a 6.5-fold higher drug permeability. These characteristics make liposome-based in situ gel an outstanding carrier for the nasal administration of donepezil HCl. Applied liposomal-based gel is expected to permeate and remain integral in the nasal mucosa by changing lipid and polar permeation pathways (70).

In vitro permeation study across sheep nasal mucosa of optimized in situ gel (batch DL 5) and donepezil HCl based in situ gel. Each value represents the mean ± SD (n=3)
Also, the hydrophilic portion of liposomes will hydrate the external surface of the nasal mucosa and help increase the acceptance of donepezil HCl by the mucosa. As the aqueous part of the liposomes enters the polar pathway, the interlamellar volume of the lipid bilayer increases, causing disturbance of the interfacial structure of the tissue (71). In addition to this, lipid components of liposomes such as cholesterol and HSPC, the nasal mucosa’s lipidic structure may also facilitate the drug uptake (72).
The stability study of the optimized batch (DL5) was carried out at 2–8°C for 6 months. As shown in Table III, an insignificant difference was observed in various parameters after 6 months of storage.
Nasal Ciliotoxicity Studies
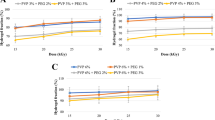
Nasal ciliotoxicity studies are used to predict the safety of the liposome ingredients. The photomicrographs of mucosa treated with phosphate buffer solution pH 6.4 (Fig. 3A) showed no nasociliary damage, the nucleus of the membrane and epithelial cell lining was visible, and the score is given as zero (normal morphology). In comparison, significant nasal mucosal damage with epithelial layer loss, nucleus loss, and mucosal layer contraction was observed in the isopropyl alcohol-treated membrane (Fig. 3B), given a score of 4 (highest tissue damage). There was no damage to the nasal mucosa and no signs of epithelial necrosis, sloughing of the epithelial cells, and hemorrhage in the membrane treated with optimized in situ gel (Fig. 3C). However, a very mild change in morphology is observed and is given a histopathological score of 1 (very mild impact). This indicated the safety of the components used in the formulation. Table IV shows a score of histopathological studies carried out using nasal mucosa. Further, all the components were used in ranges recommended in the inactive ingredient guide by the USFDA (73).

Nasal ciliotoxicity study of sheep nasal mucosa treated with A phosphate buffer solution pH 6.4, B isopropyl alcohol, C optimized donepezil liposomes based in situ gel (batch DL 5)
Preparation of Tissue Samples
The developed extraction method was sensitive for measuring the quantity of donepezil HCl in plasma and brain. The mean extraction efficiency of donepezil HCl from plasma and brain tissue homogenate was 91.02% ± 2.01% and 93.24% ± 2.14%, respectively.
Pharmacokinetic Study
In vivo pharmacokinetics has been conducted in vivo to compare donepezil HCl in plasma and brain homogenate with oral and intranasal administration of the marketed product and developed the formulations. Drug concentration versus time profiles for plasma and brain homogenate of optimized in situ gel and marketed formulation up to 8 h are shown in Fig. 4A and B, respectively. The study results showed that intranasal administration of donepezil HCl substantially altered its pharmacokinetic profile and increased its brain targeting. The AUCs of optimized in situ gel and marketed formulation following intranasal and oral administration also considerably differed. Donepezil HCl concentration in the brain post nasal administration of optimized in situ gel resulted in an AUC0→8 of 2637.27 ± 519.28 ng/ml as compared to AUC0→8 after oral administration of marketed formulation (1218.22 ± 199.53 ng/ml) (p < 0.05). These results are in line with previously published nasal administration studies that indicate enhanced bioavailability of brain drugs after intranasal administration compared to oral or parenteral administration and support the existence of a clear nose-brain pathway (44, 74, 75).

Drug concentration versus time profile in plasma and brain, A donepezil HCl liposomes–based in situ gel via intranasal administration (batch DL 5), B donepezil HCl solution via oral administration. Each data point represents the mean ± SD of six animals in each group
The optimized donepezil liposome–based in situ gel showed peak plasma concentration at 30 min (Cmax = 614.26 ± 22.09 ng/ml) and marketed formulation reached at its peak at 2 h (Cmax = 779.81 ± 32.55 ng/ml). Donepezil concentration in the brain was higher for developed in situ gel (Cmax=1239.61 ± 123.60 ng/ml), and the peak was reached at 0.5 h. In contrast, marketed formulation achieved peak concentration at 1 h with lower drug concentration (Cmax =378.12 ± 27.17 ng/ml) (Table V). The accumulation value of donepezil HCl liposome–loaded in situ gel was approximately 3.27 times higher than the marketed formulation (p < 0.05). Thus, it is evident that higher plasma concentration was achieved after oral administration while more of the drug is delivered into the brain after nasal administration. The earlier literature also reported that the nasal administration of liposomal formulation showed higher brain targeting, and the same has been observed in our study (76,77,78).
The drug targeting (DTE %) efficiency was estimated to display the percentage of donepezil HCl transmitted directly to the brain through the olfactory or trigeminal pathways. The outcomes of drug targeting efficiency were 314.29%. This could ensure the direct pathway from the nose to the brain, as suggested by Wang et al. (79).
The distribution of the donepezil HCl liposome–loaded in situ gel and marketed formulation in different organs like the heart, spleen, lungs, kidney, and liver showed significant differences, as shown in Fig. 5. The drug-loaded liposomes administered via the nasal route reached the brain via a suitable pathway, and some amount of drug enters the systemic circulation. However, the reticuloendothelial system (RES) is considered the major region of lip aggregation of liposomes after reaching the systemic circulation (80, 81). The liver, spleen, kidney, lungs, bone marrow, and lymph nodes are primary organs associated with RES. Liver has the highest liposomal uptake ability, followed by spleen, which can accumulate liposomes up to 10-fold more than other RES organs (82).

Amount of drug per gram of the brain, heart, spleen, lungs, kidney, and liver after intranasal administration (1 mg/kg) of donepezil HCl liposomes–based in situ gel (batch DL 5) and donepezil HCl solution after oral administration (1 mg/kg). The results shown as mean ± SD (n=6), significant difference (p<0.001). *p < 0.001 versus donepezil HCl solution (significant difference); #p < 0.001 versus donepezil HCl solution (no significant difference)
Liver is considered the primary organ for metabolism. Nasal administration of donepezil HCl liposome–loaded in situ gel showed 12 times lower accumulation in liver than the marketed formulation, which further supports nose to brain targeting and elimination of first-pass metabolism (83). Reduction in the distribution of the donepezil HCl in the liver minimizes its biotransformation rate and thus justifies the higher area under the curve.
The distribution of the donepezil HCl liposome–loaded in situ gel into the heart showed an insignificant difference compared to the marketed formulation. The drug concentration in the lungs and spleen showed a significant difference as more of the drug accumulated into the lungs and the spleen from the donepezil HCl liposome–loaded in situ gel (P < 0.05). The kidney is considered elimination organ for donepezil HCl; however, it might also have biliary and fecal elimination (84).
The nanosize and lipidic nature of liposomes can enable the transcellular transport of donepezil HCl through different endocytic pathways of sustentacular or neuronal cells in the olfactory membrane with higher drug concentrations in the brain (85, 86).
Biochemical Parameters
The defect in the functioning of the cholinergic system results in Alzheimer’s disease. Furthermore, the key biomarkers of cholinergic functions are AChE, SOD, GSH, catalase, and MDA. Therefore, the effects on brain homogenate levels were tested to understand the biochemical mechanism of donepezil HCl as an acetylcholinesterase inhibitor.
The acetylcholinesterase inhibitors are known to oppose scopolamine-induced amnesia (87, 88). The impact of treatment using donepezil HCl–loaded liposome-based in situ gel and marketed formulation AChE activities in the rats was evaluated. The activity of AChE was represented as OD values/mg protein, and results are shown in supplementary information Fig. S1A, The AChE levels in rats administered with scopolamine (group A) were significantly increased compared to the normal control group. The donepezil HCl–loaded liposome-based in situ gel (group B) showed a decrease in the level of AChE significantly (p < 0.05) compared to the scopolamine-induced amnesia group. The concentration of acetylcholine increased in the rat brain, and a new balance between AChE and acetylcholine could be reached by the cholinergic system, resulting in enhanced rat memory and cognitive deficits.
The scopolamine-induced amnesia group (group A) showed a significant reduction in SOD level compared to the normal control group. The administration of donepezil HCl–loaded liposome-based in situ gel significantly improved the level of SOD near the normal control group. The marketed formulation (group C) also showed an improved level of SOD compared to the scopolamine-induced amnesia group significantly (p < 0.05) (Fig. S1B).
Compared to the marketed formulation, the donepezil HCl–loaded liposome-based in situ gel showed an increased GSH in brain homogenate. However, the marketed formulation showed a substantial increase (p < 0.05) in the GSH level relative to the amnesia group caused by scopolamine (Fig. S1C).
The decrease in catalase level was observed in rats treated with a scopolamine-induced amnesia group compared to the normal control group. The catalase concentration in the rats administered with the donepezil HCl liposome-loaded in situ gel was increased to the values observed in the normal control group and thus found better than the marketed product (Fig. S1C). A significant decrease in MDA level was observed in the rats administered with donepezil HCl liposome–loaded in situ gel. It was equivalent to a normal control group and better as compared to the marketed formulation (Fig. S1D). Thus, overall, we can conclude that there is improvement in biochemical parameters post administering the developed in situ gel than the marketed product.
CONCLUSION
The donepezil liposome–based in situ gel has been prepared successfully with suitable gelling properties. It was possible to successfully formulate donepezil HCl in the form of liposomes for brain targeting via intranasal administration. The liposomes were spherical vesicles with smooth bilayer surface and size ranging from 90 to 103 nm. The optimized liposomes (DL5) showed 79% drug permeated after 480 min. The intranasal administration of formulated liposome-based in situ gel showed higher brain bioavailability than the oral administration of the marketed formulation. The pharmacokinetics showed higher Cmax, AUC0→8, and AUC0→∞ and lowered Kel in the animals administered with developed formulation over the marketed formulation. Hence, we can conclude that the developed liposome-based in situ gel can target donepezil HCl into the brain. The results of biodistribution showed that donepezil HCl liposomes based on in situ gel could be a valuable tool for brain targeting in the treatment of Alzheimer’s disease. Thus, the study concludes that the developed liposome-based in situ gel may be considered as a promising nasal delivery system for administering donepezil HCl to treat Alzheimer’s disease.
References
Alzheimer’s. Alzheimer’s disease facts and figures. Alzheimers Dement. 2020;16(3):391–460. https://doi.org/10.1002/alz.12068.
Foster ER. Themes from the special issue on neurodegenerative diseases: what have we learned, and where can we go from here? Am J Occup. 2014;68(1):6–8. https://doi.org/10.5014/ajot.2014.009910.
Wong HL, Wu XY, Bendayan R. Nanotechnological advances for the delivery of CNS therapeutics. Adv Drug Deliv Rev. 2012;64(7):686–700. https://doi.org/10.1016/j.addr.2011.10.007.
Chen D, Kay HL. Biodistribution of calcitonin encapsulated in liposomes in mice with particular reference to the central nervous system. Biochim Biophys Acta Gen Subj. 1993;1158(3):244–50. https://doi.org/10.1016/0304-4165(93)90021-Y.
Huwyler J, Wu D, Pardridge WM. Brain drug delivery of small molecules using immunoliposomes. Proc Natl Acad Sci U S A. 1996;93(24):14164–9. https://doi.org/10.1073/pnas.93.24.14164.
Rajput AP, Butani SB. Resveratrol anchored nanostructured lipid carrier loaded in situ gel via nasal route: formulation, optimization and in vivo characterization. J Drug Deliv Sci Technol. 2019;51:214–23. https://doi.org/10.1016/j.jddst.2019.01.040.
Zhang P, Chen L, Gu W, Xu Z, Gao Y, Li Y. In vitro and in vivo evaluation of donepezil-sustained release microparticles for the treatment of Alzheimer’s disease. Biomaterials. 2007;28(10):1882–8. https://doi.org/10.1016/j.biomaterials.2006.12.016.
Mistry A, Stolnik S, Illum L. Nanoparticles for direct nose-to-brain delivery of drugs. Int J Pharm. 2009;379(1):146–57. https://doi.org/10.1016/j.ijpharm.2009.06.019.
Haque S, Md S, Fazil M, Kumar M, Sahni JK, Ali J, Baboota S. Venlafaxine loaded chitosan NPs for brain targeting: pharmacokinetic and pharmacodynamic evaluation. Carbohydr Polym. 2012;89(1):72–9. https://doi.org/10.1016/j.carbpol.2012.02.051.
Salimi A, Gobadian H, Sharif MB. Dermal pharmacokinetics of rivastigmine-loaded liposomes: an ex vivo–in vivo correlation study. J Liposome Res. 2020;31:1–9. https://doi.org/10.1080/08982104.2020.1787440.
Johnsen KB, Moos T. Revisiting nanoparticle technology for blood–brain barrier transport: Unfolding at the endothelial gate improves the fate of transferrin receptor-targeted liposomes. J Control Release. 2016;222:32–46. https://doi.org/10.1016/j.jconrel.2015.11.032.
Al Asmari AK, Ullah Z, Tariq M, Fatani A. Preparation, characterization, and in vivo evaluation of intranasally administered liposomal formulation of donepezil. Drug Des Dev Ther. 2016;10:205–15. https://doi.org/10.2147/dddt.s93937.
Kanazawa T, Taki H, Okada H. Nose-to-brain drug delivery system with ligand/cell-penetrating peptide-modified polymeric nano-micelles for intracerebral gliomas. Eur J Pharm Biopharm. 2020;152:85–94. https://doi.org/10.1016/j.ejpb.2020.05.001.
Chung K, Ullah I, Kim N, Lim J, Shin J, Lee SC, Jeon S, Kim SH, Kumar P, Lee SK. Intranasal delivery of cancer-targeting doxorubicin-loaded PLGA nanoparticles arrests glioblastoma growth. J Drug Target. 2020;28(6):617–26. https://doi.org/10.1080/1061186X.2019.1706095.
Kozlovskaya L, Abou-Kaoud M, Stepensky D. Quantitative analysis of drug delivery to the brain via nasal route. J Control Release. 2014;189:133–40. https://doi.org/10.1016/j.jconrel.2014.06.053.
Agrawal M, Saraf S, Saraf S, Antimisiaris SG, Chougule MB, Shoyele SA, Alexander A. Nose-to-brain drug delivery: an update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J Control Release. 2018;281:139–77. https://doi.org/10.1016/j.jconrel.2018.05.011.
Feng Y, He H, Li F, Lu Y, Qi J, Wu W. An update on the role of nanovehicles in nose-to-brain drug delivery. Drug Discov Today. 2018;23(5):1079–88. https://doi.org/10.1016/j.drudis.2018.01.005.
Crowe TP, Greenlee MHW, Kanthasamy AG, Hsu WH. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018;195:44–52. https://doi.org/10.1016/j.lfs.2017.12.025.
Mittal D, Ali A, Md S, Baboota S, Sahni JK, Ali J. Insights into direct nose to brain delivery: current status and future perspective. Drug Deliv. 2014;21(2):75–86. https://doi.org/10.3109/10717544.2013.838713.
Doxorubicin F. Active loading method of doxorubicin hydrochloride liposomes. 2017.
Singh G, Pai RS, Pandit V. Development and validation of a HPLC method for the determination of trans-resveratrol in spiked human plasma. J Adv Pharm Technol Res. 2012;3(2):130–5. https://doi.org/10.4103/2231-4040.97296.
Wei H, Song J, Li H, Li Y, Zhu S, Zhou X, Zhang X, Yang L. Active loading liposomal irinotecan hydrochloride: preparation, in vitro and in vivo evaluation. Asian J Pharm Sci. 2013;8(5):303–11.
Al Asmari AK, Ullah Z, Tariq M, Fatani A. Preparation, characterization, and in vivo evaluation of intranasally administered liposomal formulation of donepezil. Drug design, development and therapy. 2016;10:205.
Vijayakumar MR, Kosuru R, Vuddanda PR, Singh SK, Singh S. Trans resveratrol loaded DSPE PEG 2000 coated liposomes: an evidence for prolonged systemic circulation and passive brain targeting. J Drug Deliv Sci Technol. 2016;33:125–35. https://doi.org/10.1016/j.jddst.2016.02.009.
Maritim S, Boulas P, Lin Y. Comprehensive analysis of liposome formulation parameters and their influence on encapsulation, stability and drug release in glibenclamide liposomes. Int J Pharm. 2021;592:120051. https://doi.org/10.1016/j.ijpharm.2020.120051.
Swami R, Kumar Y, Chaudhari D, Katiyar SS, Kuche K, Katare PB, Banerjee SK, Jain S. pH sensitive liposomes assisted specific and improved breast cancer therapy using co-delivery of SIRT1 shRNA and docetaxel. Mater Sci Eng C. 2021;120:111664. https://doi.org/10.1016/j.msec.2020.111664.
Abdelbary G. Ocular ciprofloxacin hydrochloride mucoadhesive chitosan-coated liposomes. Pharm Dev Technol. 2011;16(1):44–56.
Kassem MA, Aboul-Einien MH, El Taweel MM. Dry gel containing optimized felodipine-loaded transferosomes: a promising transdermal delivery system to enhance drug bioavailability. AAPS PharmSciTech. 2018;19(5):2155–73.
Maestrelli F, González-Rodríguez ML, Rabasco AM, Mura P. Preparation and characterisation of liposomes encapsulating ketoprofen–cyclodextrin complexes for transdermal drug delivery. Int J Pharm. 2005;298(1):55–67.
Gabal YM, Kamel AO, Sammour OA, Elshafeey AH. Effect of surface charge on the brain delivery of nanostructured lipid carriers in situ gels via the nasal route. Int J Pharm. 2014;473(1):442–57. https://doi.org/10.1016/j.ijpharm.2014.07.025.
Pachis K, Blazaki S, Tzatzarakis M, Klepetsanis P, Naoumidi E, Tsilimbaris M, Antimisiaris SG. Sustained release of intravitreal flurbiprofen from a novel drug-in-liposome-in-hydrogel formulation. Eur J Pharm Sci. 2017;109:324–33. https://doi.org/10.1016/j.ejps.2017.08.028.
Phaechamud T, Mahadlek J. Solvent exchange-induced in situ forming gel comprising ethyl cellulose-antimicrobial drugs. Int J Pharm. 2015;494(1):381–92.
Masiuk T, Kadakia P, Wang Z. Development of a physiologically relevant dripping analytical method using simulated nasal mucus for nasal spray formulation analysis. J Pharm Anal. 2016;6(5):283–91.
Cai Z, Song X, Sun F, Yang Z, Hou S, Liu Z. Formulation and evaluation of in situ gelling systems for intranasal administration of gastrodin. AAPS PharmSciTech. 2011;12(4):1102–9. https://doi.org/10.1208/s12249-011-9678-y.
Cai Z, Song X, Sun F, Yang Z, Hou S, Liu Z. Formulation and evaluation of in situ gelling systems for intranasal administration of gastrodin. AAPS PharmSciTech. 2011;12(4):1102–9.
de Souza IFF, dos Santos TQ, Placido RV, Mangerona BA, Carvalho FC, Boralli VB, Ruela ALM, Pereira GR. The liquid crystalline phase behaviour of a nasal formulation modifies the brain disposition of donepezil in rats in the treatment of Alzheimer’s disease. Colloids Surf B: Biointerfaces. 2021;203:111721. https://doi.org/10.1016/j.colsurfb.2021.111721.
Morsi N, Ghorab D, Refai H, Teba H. Ketoroloac tromethamine loaded nanodispersion incorporated into thermosensitive in situ gel for prolonged ocular delivery. Int J Pharm. 2016;506(1-2):57–67.
Shah B, Khunt D, Bhatt H, Misra M, Padh H. Intranasal delivery of venlafaxine loaded nanostructured lipid carrier: risk assessment and QbD based optimization. J Drug Deliv Sci Technol. 2016;33:37–50.
Naik A, Nair H. Formulation and evaluation of thermosensitive biogels for nose to brain delivery of doxepin. Biomed Res Int. 2014;2014:847547. https://doi.org/10.1155/2014/847547.
Sood S, Jain K, Gowthamarajan K. Optimization of curcumin nanoemulsion for intranasal delivery using design of experiment and its toxicity assessment. Colloids Surf B: Biointerfaces. 2014;113:330–7. https://doi.org/10.1016/j.colsurfb.2013.09.030.
Ali J, Ali M, Baboota S, Ali R, Mittal G, Bhatnagar A. Reflection on existence of neural and non-neural pathway for nose-to-brain using a novel formulation of an anticholinesterase piperidine derivative. Curr Nanosci. 2010;6(3):320–3.
Barnett SW. Manual of animal technology2007.
Al-Subaie MM, Hosny KM, El-Say KM, Ahmed TA, Aljaeid BM. Utilization of nanotechnology to enhance percutaneous absorption of acyclovir in the treatment of herpes simplex viral infections. Int J Nanomedicine. 2015;10:3973.
Sherry Chow HH, Chen Z, Matsuura GT. Direct transport of cocaine from the nasal cavity to the brain following intranasal cocaine administration in rats. J Pharm Sci. 1999;88(8):754–8. https://doi.org/10.1021/js9900295.
Abdelbary GA, Tadros MI. Brain targeting of olanzapine via intranasal delivery of core–shell difunctional block copolymer mixed nanomicellar carriers: in vitro characterization, ex vivo estimation of nasal toxicity and in vivo biodistribution studies. Int J Pharm. 2013;452(1):300–10. https://doi.org/10.1016/j.ijpharm.2013.04.084.
Panchal SS, Patidar RK, Jha AB, Allam AA, Ajarem J, Butani SB. Anti-inflammatory and antioxidative stress effects of oryzanol in glaucomatous rabbits. J Ophthalmol. 2017;2017:1468716. https://doi.org/10.1155/2017/1468716.
Šinko G, Čalić M, Bosak A, Kovarik Z. Limitation of the Ellman method: cholinesterase activity measurement in the presence of oximes. Anal Biochem. 2007;370(2):223–7. https://doi.org/10.1016/j.ab.2007.07.023.
Kovaceva J, Pláteník J, Vejrazka M, Stípek S, Ardan T, Cejka C, Midelfart A, Cejková J. Differences in activities of antioxidant superoxide dismutase, glutathione peroxidase and prooxidant xanthine oxidoreductase/xanthine oxidase in the normal corneal epithelium of various mammals. Physiol Res. 2007;56(1):105–12.
Mohandas J, Marshall JJ, Duggin GG, Horvath JS, Tiller DJ. Differential distribution of glutathione and glutathione-related enzymes in rabbit kidney: possible implications in analgesic nephropathy. Biochem Pharmacol. 1984;33(11):1801–7. https://doi.org/10.1016/0006-2952(84)90353-8.
Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121–6. https://doi.org/10.1016/s0076-6879(84)05016-3.
Grotto D, Maria LS, Valentini J, Paniz C, Schmitt G, Garcia SC, Pomblum VJ, Rocha JBT, Farina M. Importance of the lipid peroxidation biomarkers and methodological aspects for malondialdehyde quantification. Quim Nova. 2009;32(1):169–74.
Narayan R, Singh M, Ranjan O, Nayak Y, Garg S, Shavi GV, Nayak UY. Development of risperidone liposomes for brain targeting through intranasal route. Life Sci. 2016;163:38–45. https://doi.org/10.1016/j.lfs.2016.08.033.
Chavan SS, Ingle SG, Vavia PR. Preparation and characterization of solid lipid nanoparticle-based nasal spray of budesonide. Drug Deliv Transl Res. 2013;3(5):402–8. https://doi.org/10.1007/s13346-012-0105-z.
Chen M, Liu X, Fahr A. Skin penetration and deposition of carboxyfluorescein and temoporfin from different lipid vesicular systems: in vitro study with finite and infinite dosage application. Int J Pharm. 2011;408(1):223–34. https://doi.org/10.1016/j.ijpharm.2011.02.006.
Badran M. Formulation and in vitro evaluation of flufenamic acid loaded deformable liposomes for improved skin delivery. Digest J Nanomater Biostruct (DJNB). 2014;9(1).
Putri DCA, Dwiastuti R, Marchaban M, Nugroho AK. Optimization of mixing temperature and sonication duration in liposome preparation. J Pharm Sci Commun. 2017;14(2):79–85.
Agrawal M, Tripathi DK, Saraf S, Saraf S, Antimisiaris SG, Mourtas S, et al. Recent advancements in liposomes targeting strategies to cross blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. J Control Release. 2017;260:61–77.
Barbălată CI, Porfire AS, Sesarman A, Rauca V-F, Banciu M, Muntean D, Știufiuc R, Moldovan A, Moldovan C, Tomuță I. A screening study for the development of simvastatin-doxorubicin liposomes, a co-formulation with future perspectives in colon cancer therapy. Pharmaceutics. 2021;13(10):1526.
Gubernator J. Active methods of drug loading into liposomes: recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin Drug Deliv. 2011;8(5):565–80.
Kita K, Dittrich C. Drug delivery vehicles with improved encapsulation efficiency: taking advantage of specific drug–carrier interactions. Expert Opin Drug Deliv. 2011;8(3):329–42.
Sur S, Fries AC, Kinzler KW, Zhou S, Vogelstein B. Remote loading of preencapsulated drugs into stealth liposomes. Proc Natl Acad Sci. 2014;111(6):2283–8.
Zhang W, Wang G, Falconer JR, Baguley BC, Shaw JP, Liu J, Xu H, See E, Sun J, Aa J, Wu Z. Strategies to maximize liposomal drug loading for a poorly water-soluble anticancer drug. Pharm Res. 2015;32(4):1451–61.
Barenholz Y. Relevancy of drug loading to liposomal formulation therapeutic efficacy. J Liposome Res. 2003;13(1):1–8.
Vakili-Ghartavol R, Rezayat SM, Faridi-Majidi R, Sadri K, Jaafari MR. Optimization of docetaxel loading conditions in liposomes: proposing potential products for metastatic breast carcinoma chemotherapy. Sci Rep. 2020;10(1):5569. https://doi.org/10.1038/s41598-020-62501-1.
Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta Biomembr. 1993;1151(2):201–15. https://doi.org/10.1016/0005-2736(93)90105-9.
Schwendener RA, Schott H. Liposome formulations of hydrophobic drugs. Methods Mol Biol (Clifton, NJ). 2010;605:129-38. https://doi.org/10.1007/978-1-60327-360-2_8.
Jia LJ, Zhang DR, Li ZY, Feng FF, Wang YC, Dai WT, Duan CX, Zhang Q. Preparation and characterization of silybin-loaded nanostructured lipid carriers. Drug Deliv. 2010;17(1):11–8. https://doi.org/10.3109/10717540903431586.
S-l C, X-w R, Q-z Z, Chen E, Xu F, Chen J, et al. In situ gel based on gellan gum as new carrier for nasal administration of mometasone furoate. Int J Pharm. 2009;365(1):109–15. https://doi.org/10.1016/j.ijpharm.2008.08.042.
Pathan IB, Chudiwal V, Farooqui I, Shingare P. Formulation design and evaluation of nasal in situ gel as a novel vehicle for azelastine hydrochloride. Int J Drug Deliv. 2013;5(3):284.
Yong CS, Choi JS, Quan Q-Z, Rhee J-D, Kim C-K, Lim S-J, Kim KM, Oh PS, Choi HG. Effect of sodium chloride on the gelation temperature, gel strength and bioadhesive force of poloxamer gels containing diclofenac sodium. Int J Pharm. 2001;226(1-2):195–205.
Hosny KM. Preparation and evaluation of thermosensitive liposomal hydrogel for enhanced transcorneal permeation of ofloxacin. AAPS PharmSciTech. 2009;10(4):1336–42. https://doi.org/10.1208/s12249-009-9335-x.
Hong SS, Oh KT, Choi HG. Liposomal formulations for nose-to-brain delivery: recent advances and future perspectives. 2019;11(10). https://doi.org/10.3390/pharmaceutics11100540.
Hashemzadeh S, Hashemzadeh K, Ranjbari A, Halimi M, Estakhri R, Aligholipour R, et al. Erythropoietin does not ameliorate limb schemia/reperfusion injury in rabbits. Afr J Pharm Pharmacol. 2012;6(20):1457–61.
Varshosaz J, Sadrai H, Heidari A. Nasal delivery of insulin using bioadhesive chitosan gels. Drug Deliv. 2006;13(1):31–8. https://doi.org/10.1080/10717540500309040.
Basu S, Chakrabatorty S, Bandyopadhyay AK. Development and evaluation of a mucoadhesive nasal gel of midazolam prepared with Linum usitatissimum L. seed mucilage. Sci Pharm. 2009;77(4):899–910.
Afergan E, Epstein H, Dahan R, Koroukhov N, Rohekar K, Danenberg HD, Golomb G. Delivery of serotonin to the brain by monocytes following phagocytosis of liposomes. J Control Release. 2008;132(2):84–90. https://doi.org/10.1016/j.jconrel.2008.08.017.
Lv W, Guo J, Li J, Huang L, Ping Q. Distribution of liposomal breviscapine in brain following intravenous injection in rats. Int J Pharm. 2005;306(1):99–106. https://doi.org/10.1016/j.ijpharm.2005.09.012.
Xiang Y, Long Y, Yang Q, Zheng C, Cui M, Ci Z, Lv X, Li N, Zhang R. Pharmacokinetics, pharmacodynamics and toxicity of Baicalin liposome on cerebral ischemia reperfusion injury rats via intranasal administration. Brain Res. 1726;2020:146503. https://doi.org/10.1016/j.brainres.2019.146503.
Wang F, Jiang X, Lu W. Profiles of methotrexate in blood and CSF following intranasal and intravenous administration to rats. Int J Pharm. 2003;263(1):1–7. https://doi.org/10.1016/S0378-5173(03)00341-7.
Senior J. Fate and behavior of liposomes in vivo: a review of controlling factors. Crit Rev Ther Drug Carrier Syst. 1987;3(2):123–93.
Poste G, Papahadjopoulos D, Vail WJ. Lipid vesicles as carriers for introducing biologically active materials into cells. Methods Cell Biol: Elsevier. 1976:33–71.
Chrai SS, Murari R, Ahmad I. Liposomes (a review). Part two: Drug delivery systems. BioPharm. 2002;15(1).
Tiseo PJ, Perdomo CA, Friedhoff LT. Metabolism and elimination of 14C-donepezil in healthy volunteers: a single-dose study. Br J Clin Pharmacol. 1998;46(Suppl 1):19–24. https://doi.org/10.1046/j.1365-2125.1998.0460s1019.x.
Matsui K, Mishima M, Nagai Y, Yuzuriha T, Yoshimura T. Absorption, distribution, metabolism, and excretion of donepezil (Aricept) after a single oral administration to rat. Drug Metab Dispos. 1999;27(12):1406–14.
Ikechukwu Ugwoke M, Kaufmann G, Verbeke N, Kinget R. Intranasal bioavailability of apomorphine from carboxymethylcellulose-based drug delivery systems. Int J Pharm. 2000;202(1):125–31. https://doi.org/10.1016/S0378-5173(00)00434-8.
Devkar TB, Tekade AR, Khandelwal KR. Surface engineered nanostructured lipid carriers for efficient nose to brain delivery of ondansetron HCl using Delonix regia gum as a natural mucoadhesive polymer. Colloids Surf B: Biointerfaces. 2014;122:143–50. https://doi.org/10.1016/j.colsurfb.2014.06.037.
Chen J, Long Y, Han M, Wang T, Chen Q, Wang R. Water-soluble derivative of propolis mitigates scopolamine-induced learning and memory impairment in mice. Pharmacol Biochem Behav. 2008;90(3):441–6. https://doi.org/10.1016/j.pbb.2008.03.029.
Lima JA, Costa RS, Epifânio RA, Castro NG, Rocha MS, Pinto AC. Geissospermum vellosii stembark: anticholinesterase activity and improvement of scopolamine-induced memory deficits. Pharmacol Biochem Behav. 2009;92(3):508–13. https://doi.org/10.1016/j.pbb.2009.01.024.
Acknowledgements
The authors are thankful to Dr. Alex George, Vice President, Dr. Reddy’s Laboratory, Hyderabad, India, and Mr. Shushrut Kulkarni, Vice President, Glenmark Pharmaceutical Limited, Mumbai, India, for granting permission during their tenure to work in Zydus Healthcare Limited, PTC, Moraiya, Ahmedabad, India. The authors are thankful to Nikita Gupta and Ruchi Sawhney for language modification, grammar, and typographic corrections.
Author information
Authors and Affiliations
Contributions
Dr. Amarjitsing Rajput: formulation development, analysis, investigation, and writing: original draft. Dr. Shital Butani: concept formulation, supervision, editing, and review
Corresponding author
Ethics declarations
This article does not contain any studies in humans performed by any of the authors.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
Biochemical parameter study. A Effect on the level of AChE B) Effect on the level of SOD C) Effect on the level of GSH D) Effect on the level of Catalase E) Effect on the level of MDA. Statistical analysis was carried out using one-way ANOVA followed by Tukey’s multiple comparison test; values are statistically significant at *p < 0.05 versus Normal control; #p < 0.05 versus optimized in situ gel. Group A. Normal control. Group B. Optimized in situ gel (Batch DL 5). Group C. Donepezil HCl solution (JPG 120 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Rajput, A., Butani, S. Donepezil HCl Liposomes: Development, Characterization, Cytotoxicity, and Pharmacokinetic Study. AAPS PharmSciTech 23, 74 (2022). https://doi.org/10.1208/s12249-022-02209-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-022-02209-9