
Abstract
The postprandial glycemic regulation is essential for diabetic patients to reduce the risk of long-term microvascular and macrovascular complications. Herein, we designed a glucose-responsive oral insulin delivery system based on polyelectrolyte complexes (PECs) for controlling the increasing postprandial glucose concentrations. Briefly, alginate-g-3-aminophenylboronic acid (ALG-g-APBA) and chitosan-g-3-fluoro-4-carboxyphenylboronic acid (CS-g-FPBA) were wrapped on mesoporous silica (MSN) to form the negative charged ALG-g-APBA@MSN and the positive charged CS-g-FPBA@MSN nanoparticles, with an optimum insulin loading capacity of 124 mg/g and 295 mg/g, respectively. ALG-g-APBA@MSN was further cross-linked with CS-g-FPBA@MSN to form PECs through electrostatic interaction and borate esters. The dense polyelectrolyte network wrapped on MSN was capable of preventing insulin from diffusion and regulating its release. The in vitro insulin release of PECs demonstrated an obvious glucose response profile in different glucose concentrations (0 mg/mL, 2 mg/mL, 5 mg/mL) and presented a switch “on” and “off” release regulation at hyperglycemic or normal state. The CCK-8 assay showed that none of the MSN, ALG-g-APBA@MSN, CS-g-FPBA@MSN, and PECs possessed cytotoxicity to Caco-2 cells. For in vivo tests, the oral PECs exhibited a significant hypoglycemic effect and maintained in the euglycemic levels up to approximately 12 h on diabetic rats. Overall, the PECs directly triggered by postprandial glucose in the intestine have a good potential to be applied in intelligent insulin delivery by the oral route.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Achieving tight control of blood glucose levels is the main medical management of diabetes, for preventing the long-term organ damage caused by chronic hyperglycemia [1]. The traditional treatment is frequent subcutaneous injection of insulin, according to carbohydrate content before or after meals to mitigate postprandial glycemic excursions [2]. However, this treatment is usually accompanied with some problems such as pains, hypoglycemia, and glucose fluctuation due to the difficulty of dose control. The blood glucose fluctuation may lead to severe complications and chronic organ damages to the nervous system, heart, kidney, eyes, or skin [3]. Thereby, a noninvasive and intelligent delivery system that combines postprandial glycemic monitoring with self-regulation of insulin release is urgently required.
Various noninvasive administrations have become the research focus in developing insulin delivery systems such as nasal, lung, microarray patch, and microneedles [4]. Those strategies are currently not optimal for drug delivery of insulin considering their low penetration, indeterminate biological safety, cilia scavenging effect, and inconvenient management. For safety and high patient compliance, oral insulin delivery platforms emerged as nanoparticles, liposomes, microspheres, and polyelectrolyte complexes present encouraging prospects these years [5,6,7,8]. Nevertheless, this administration has several drawbacks, such as enzymatic degradation, poor drug absorption, and short residence time in the intestinal tract. To overcome these hurdles, the oral polyelectrolyte complexes (PECs) formed by natural polymers of chitosan and alginate were reported as a promising insulin delivery system according to our previous and other’s work [9]. Chitosan, as a natural biopolymer with good biocompatibility, adhesion, nontoxicity, and biodegradability, has been widely applied in the research of biomedicine and drug delivery [10, 11]. More importantly, chitosan can facilitate the transport of protein and peptides via opening tight junctions between epithelial cells [12,13,14,15]. Alginate also serves as a desirable candidate in oral administration for its ability to shrink under gastric environment conditions, thereby protecting the loaded drugs from acid [16, 17].
Compared with these organic biopolymers, some inorganic materials especially mesoporous silica nanoparticles (MSN) are regarded as an ideal drug container for the advantages including good stability, large surface area, tunable pore size, uniform morphology, and ease of functionalization [18]. Orally administered silica nanoparticles can be easily excreted or degraded into silicic acid, which is the most bioavailable form of silicon [19]. MSN not only present more effective protection for macromolecules in the gastric environment but facilitate permeation in the intestine as well [18, 20]. Moreover, modification or coating on MSN can achieve drug release based on stimuli and, thus, provide better control of drug release [21,22,23].
Importantly, how to further realize the intelligent insulin release on demand needs to be considered for MSN. The intelligent glucose-responsive delivery system is advantageous to self-regulate insulin release according to different glucose levels and avoid the risks associated with frequent subcutaneous injection. The related functional sections in the glucose stimuli system including phenylboronic acid (PBA) [24], glucose oxidase (GOx) [25], and glucose-binding proteins (Con A) [26], while the first artificial compounds PBA usually perform better in structural stability and storage than the others [27, 28]. PBA could exist as the uncharged trigonal hydrophobic state or the negatively charged tetrahedral hydrophilic state in an aqueous medium. The latter forms reversible ester bonds with 1,2-diol and 1,3-diol compounds to enhance its hydrophilicity [29, 30]. However, what makes the PBA less sensitive to glucose in physiological conditions is its high pKa value (8.2–8.6) [31]. Some researchers have tried to reduce the pKa of PBA by introducing electron-withdrawing groups (-COOH, -F, -Cl, -NO2) [24, 32].
Herein, we designed a noninvasive and intelligent glucose-responsive delivery system with a smart release switch for glucose regulation. The nano-carrier was composed of inorganic MSN as a stable encapsulant for insulin and biopolymers as a coating layer. MSN was separately coated with alginate-g-3-aminophenylboronic acid (ALG-g-APBA) and chitosan-g-3-carboxy-4-fluorophenylboronic acid (CS-g-FPBA) to fabricate the negatively charged ALG-g-APBA@MSN-NH2 and the positively charged CS-g-FPBA@MSN, after which the oppositely charged nanoparticles cross-linked to form PECs through electrostatic interaction (Scheme 1, 2). Simultaneously, diol moieties on ALG-g-APBA could bind with boronic acid of CS-g-FPBA to form borate esters, and diol moieties of CS-g-FPBA bind to PBA of ALG-g-APBA. The biopolymer network coated on MSN associated with electrostatic interaction and borate esters acted as an intelligent monitor to control insulin release, swollen after glucose competitively bond to PBA on biopolymers in high glucose condition and shrunk following biopolymers replaced glucose to recombine with PBA at glucose-absence medium. Moreover, the network as well as MSN can protect insulin from diffusion in the gastric environment and improve intestinal permeability. The pH and glucose responsiveness of PECs in vitro were investigated, as well as its switch “on” and “off” regulation on release. In addition, the cytotoxicity was measured by the CCK-8 method. Finally, the postprandial hypoglycemic effect of PECs was evaluated on diabetic mice.

Synthesis of ALG-g-APBA (a) and CS-g-FPBA (b) using the principles of carbodiimide chemistry

Schematic representation for fabrication and glucose-responsive release of PECs
MATERIALS AND METHODS
Materials
Cetyltrimethylammonium chloride (CTAC, purity>95%), triethanolamine (TEA), tetraethyl orthosilicate (TEOS), and 3-amionpropyltriethoxysilane (APTES) were all obtained from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). Chitosan (degree of deacetylation>95%), sodium alginate, 3-aminophenylboronic acid (APBA, purity>95%), 3-carboxy-4-fluorophenylboronic acid (FPBA, purity>95%), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropylcarbodiimide) hydrochloride (EDC), and deuterium oxide (D2O) were all purchased from Aladdin Industrial Corporation (Shanghai, China). Porcine insulin (INS, purity>98%) was purchased from Xuzhou Wanbang Jinqiao Pharmaceutical Co., Ltd (Jiangsu, China). Bicinchoninic Acid Protein Assay Kit (BCA) and Bradford Protein Assay Kit were both purchased from Beyotime Biotechnology Co., Ltd (Shanghai, China). The cell counting kit-8 (CCK-8) was obtained from Dojindo Laboratories (Tokyo, Japan). The glucose meter (ACCU-CHEK Active) was purchased from Roche Diagnostics GmbH (Basel, Switzerland). Porcine Insulin ELISA Kit was obtained from Shanghai Enzyme-linked Biotechnology Co., Ltd (Shanghai, China). Male Sprague-Dawley (SD) rats were available from the Experimental Animal Center of Southern Medical University (Guangzhou, China).
Characterization
Proton nuclear magnetic resonance (1H-NMR spectrums, AVANCE III 400 MHz, Bruker, Germany) of the polymers were characterized to confirm structure in solvent of D2O. Fourier transform infrared (FTIR) spectrums were tested in KBr pellets with Fourier transform infrared spectrometer (Nicolet 6700, Thermo Fisher, USA) in the wavelength of 300–4000 cm-1. Morphology images were carried out with a transmission electron microscope (TEM, JEM-2100F, JEOL, Japan) by dispersing nanoparticles in deionized water. Particle size and zeta potential (Zetasizer Nano ZS, Malvern, UK) were measured by nanoparticles dispersing in PBS buffer (pH =7.4). The nitrogen adsorption-desorption isotherms (TristarII 3020, Micromeritics, USA) were characterized to properties of mesoporous materials. The entrapment efficiency (EE), loading capacity (LE), and cumulative release profiles were recorded with a microplate reader (Infinite M1000 Pro, Tecan, Switzerland) in a 96-well plate.
Fabrication of PECs
Synthesis of Dendritic Mesoporous Silica (MSN)
The 3D-dendritic mesoporous silica was prepared by a one-pot biphase stratification approach [33, 34]. Briefly, 24 mL of CTAC solution (10 wt% ) is mixed with 66 μL of TEA and 36 mL of H2O as water phase and magnetically stirred gently for 1 h at 70°C. Then, 20 mL of TEOS in cyclohexane(20 v/v%) as oil phase was added to the prepared water phase and stirred 12 h at a speed of 200 rpm at 70°C. After removing the upper oil phase, the product was centrifuged (12,000 rpm, 20 min) and washed with ethanol and ultrapure water. In order to remove the template, the precipitate after centrifugation was dispersed in a mixture of 150-ml absolute ethanol and 1.5-ml hydrochloric acid and refluxed for 24 h at 80°C. The product was centrifuged (12,000 rpm, 5 min), washed three times with ethanol, and dried at 100°C for 6 h.
To modify amino groups on the MSN, 150 mg of MSN and 200 μL of APTES were dispersed in 60 ml of ethanol and refluxed for 24 h at 80°C. The amino-functionalized MSN was collected via centrifugation (12,000 rpm, 5 min) and washed three times with ethanol.
Synthesis of Alginate-g-3-Aminophenylboronic Acid (ALG-g-APBA)
Alginate-g-3-aminophenylboronic acid was synthesized via coupling agents as illustrated in Scheme 1a [35, 36]. Alginate (1 g) dissolved in 100 mL of ultrapure water overnight was mixed with 0.96 g of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and 0.575 g N-hydroxysuccinimide (NHS) as coupling agents to activate the carboxyl groups for 45 min. While the pH was adjusted to 5.5 with 1-M NaOH and stirred for 24 h at ambient temperature, 0.78 g of 3-aminophenylboronic acid (3-APBA) dissolved in 1 mL of methanol was added to the activated solution. The reaction product was dialyzed against ultrapure water for 3 days (MWCO=3,500 Da) and lyophilized.
Synthesis of Chitosan-g-3-Fluoro-4-Carboxyphenylboronic Acid (CS-g-FPBA)
Chitosan-g-3-fluoro-4-carboxyphenylboronic acid (CS-g-FPBA) was synthesized by the same method with ALG-g-APBA [37]. As shown in Scheme 1b, 0.12 g of chitosan (CS) was dissolved in 20 mL of 1% (v/v) acetic acid solution with 170 mL of PBS buffer. Added 20 mL of dimethyl sulfoxide (DMSO) in the flask with 0.275-g 4-fluoro-3-carboxyphenylboronic acid (FPBA), 0.288-g 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), and 0.172-g N-hydroxysuccinimide (NHS), the mixture was stirred and activated for 4 h. Then the activated product was added to the previously dissolved chitosan solution, and the pH was adjusted to 12.0 with 1-M NaOH and stirred for 24 h at room temperature. After the reaction, the product was purged by dialyzing against with 10% (v/v) ethanol solution and ultrapure water for 3 days (MWCO=12,000 Da) and freeze-dried.
Preparation of ALG-g-APBA-Coated MSN-NH2(INS) Nanoparticles (ALG-g-APBA@MSN)
The nanoparticles were prepared by solvent adsorption method as previously reported [38]. Briefly, the ALG-g-APBA solution (1 mg/ml) was prepared by dissolving ALG-g-APBA in phosphate buffer (pH=7.4). Then, insulin powder dissolved in 0.01-M HCl was stirred with MSN-NH2 for 2 h in an ice bath whereas the pH value was adjusted to 6.0. The insulin-loaded MSN-NH2 nanoparticle suspension was added dropwise to the ALG-g-APBA solution (the optimal mass ratio of INS:MSN-NH2:ALG-g-APBA=2:2:1), stirred for 2 h. ALG-g-APBA@MSN nanoparticles were obtained after centrifugation (10,000 rpm, 5 min, 4°C) and washed twice with PBS buffer (pH=5.3). The free insulin in the supernatant was determined by BCA protein assay with a standard curve of a different concentration at 562 nm [39]. The entrapment efficiency (EE) and loading capacity (LE) rate were calculated as:
where weighttotal is the total amount of insulin and weightfree is the free insulin in the supernatant solution:
where weighttotal is the total amount of insulin, weightfree is the free insulin in the supernatant solution, weightnanoparticle is the weight of nanoparticles.
Preparation of CS-g-FPBA-Coated MSN (INS) Nanoparticles (CS-g-FPBA@MSN)
CS-g-FPBA was dissolved in phosphate buffer (pH=4.0) with a concentration of 1 mg/mL. Rh-insulin dissolved in 0.01-M HCl (2 mg/mL) was stirred with MSN powder for 2 h in an ice bath. The insulin-loaded MSN nanoparticle suspension was added dropwise to the CS-g-FPBA solution and stirred for 2 h. CS-g-FPBA@MSN nanoparticles were collected by centrifugation (10,000 rpm, 5 min, 4°C) and washed twice with PBS buffer (pH=5.3). The entrapment efficiency (EE) and loading capacity (LE) rate were calculated as previous equations.
Fabrication of Polyelectrolyte Complexes (PECs)
ALG-g-APBA@MSN and CS-g-FPBA@MSN were separately dispersed in PBS buffer (pH=5.3). CaCl2 solution, 200 μL, (5 mg/mL) was mixed with ALG-g-APBA@MSN solution, after which CS-g-FPBA@MSN solution was dropwise added and stirred for 1 h [40]. The PECs were obtained by centrifugation (10,000 rpm, 5 min, 4°C), washed with PBS buffer, and lyophilized. The entrapment efficiency (EE) and loading capacity (LE) rates were calculated via the same method in the previous equations.
In Vitro Release Studies
The pH and glucose sensitivity of PECs were investigated by dispersing them in different media (pH=1.2, pH=7.4, and glucose=0, 2, 5 mg/mL) using a constant temperature shaker (100 rpm) at 37°C [41]. At predetermined time points (1, 2, 3, 4, 5, 6, 8, 10, 12, 24 h), the suspension was centrifuged (6,000 rpm, 1 min) and 0.2 mL of supernatant was taken out for measurement. Then, 0.2 mL of fresh solution with the same pH value and glucose concentration was supplied to the remaining suspension. All the experiments were performed in triplicate. The collected supernatant was estimated via the Bradford protein assay by a UV spectrometer at 595 nm, and the cumulatively released amount under different pH values and glucose concentrations was calculated [39].
The capacity of switch “on” and “off” release was estimated through transferring the PECs in a glucose solution and glucose-absence solution. In brief, the nanoparticles were dispersed and shook gently in glucose solution (5 mg/mL) at 37°C. After 3 h, the suspension was centrifuged (6,000 rpm, 1 min, 4°C), and the supernatant was removed. The precipitate was dispersed in PBS buffer without glucose to shake for another 3 h. The nanoparticles after centrifugation were redispersed in glucose solution (5 mg/mL) and PBS buffer for a new cycle. The concentrations of the sample were determined through the Bradford method at 595 nm for calculating the cumulative release rate under different conditions.
Cell Viability
The cell counting kit-8 (CCK-8) was used to estimate the cell viability of blank nanoparticles on Caco-2 cells as previously reported [42, 43]. The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; high glucose) with 10% (v/v) fetal bovine serum at a humidified atmosphere (37°C, 5% CO2). The Caco-2 cells in the logarithmic growth phase which proliferated to cover the bottle wall were seeded onto 96-well plates at a density of 5×103 cells per well with 100 μl of growth medium containing 10% FBS. After the cells grow against the wall of flask, the original culture medium was discarded, and the culture medium was separately replaced with ALG-g-APBA@MSN solution and CS-g-FPBA@MSN solution in a range of concentrations from 15.625 μg/mL to 1000 μg/mL (15.625, 31.25, 62.5, 125, 250, 500, 1000 μg/mL). Cells without the treatment of nanoparticles were used as the control group whose viability was set as 100%. Each concentration was performed in 5 duplicate wells. After interacted with the cells for 24 h, the solution was discarded and washed with PBS buffer. Added to each well was 100 μL of 10% CCK-8 solution and incubated at 37°C for 4 h. The optical density values were tested at the absorbance of 450 nm, and then the cell viability was calculated as follows:
In Vivo Release Studies
Healthy male Sprague-Dawley (SD) rats were provided from the Experimental Animal Center of Southern Medical University (Guangzhou, China), and the rules of the Institutional Animal Care and Ethics Committee of Southern Medical University were followed. The rats were given standard dehydrated granules and water as well kept at a standard temperature (25 ±2°C) under an alternating 12-h light-dark cycle. All animal experiments are conducted in full compliance with local, national, ethical, and regulatory principles and local licensing regulations, per the spirit of Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International’s expectations for animal care and use/ethics committees.
The hypoglycemic efficiency of insulin-loaded oral nanoparticles was investigated on five healthy rats and twenty diabetic male rats induced by intraperitoneal injection of streptozotocin (STZ, in 10-mM citrate buffer, pH 4.5) at a dose of 60 mg/kg [44]. The fasting blood glucose was measured 1 week after establishing the model. The rats were considered diabetic if the fasting blood glucose value was over 16.7 mmol/L for 3 days consecutively [45]. All the rats (180–220 g) were on a fast overnight whereas allowed for water and randomly divided into four groups (A, B, C, D, E, n=5). Group A was injected subcutaneously with insulin solution (5 IU/kg), group B (PECs; 50 IU/kg), group C (PECs without PBA modification, 50 IU/kg), and group D (insulin solution; 50 IU/kg) were orally given by intragastric gavage. PECs without PBA modification nanoparticles were polyelectrolyte complexes composed of alginate-coated MSN-NH2 nanoparticles and chitosan-coated MSN nanoparticles. Group E were normal rats given PECs (50 IU/kg) through gavage as well. After 30 min, all the experiment groups were further orally administered glucose solution (1 g/kg) to simulate postprandial glucose levels. The blood glucose was monitored at predetermined time points using a glucometer (Roche). To measure the insulin concentrations of diabetic rats, blood samples (500 μL) were collected from orbit at determined time intervals and centrifuged (3,500 rpm, 10 min). Insulin contents in serum were determined by an insulin ELISA (enzyme-linked immunosorbent assays) kit. The area below the curve of plasma insulin vs. time (AUC) was calculated using the trapezoidal role, and the relative bioavailability (F%) of oral PECs was calculated in the following equation:
Statistical Analyses
Statistical analyses were carried out with IBM SPSS Statistics 24. Independent student’s t test and descriptive statistic were utilized to access significant difference between groups. A significant difference could be indicated when p<0.05.
RESULT AND DISCUSSION
Fabrication and Characterization of Polyelectrolyte Complexes
Modification and Characterization of ALG-g-APBA and CS-g-FPBA
The FTIR spectrum of mesoporous silica (MSN) is shown in Figure 1a, the strong and broad absorbance band of 1,000 cm-1 was antisymmetric, and stretching vibration peaks of Si-O-Si and the peaks at 963 cm-1 and 800 cm-1 corresponded to Si-O stretching vibration [46]. The structure of ALG-g-APBA was characterized with the 1H-NMR spectrum shown in Figure 2b. The resonance peaks at δ=3.5–4.8 were attributed to protons of carbon skeleton in alginate [38]. The peaks at δ=7.2–7.8 corresponded to protons of benzene ring on phenylboronic acid, indicating phenylboronic acid has been successfully grafted onto the glucosamine units of alginate. All the information was consistent with the results of the FTIR spectrum shown in Figure 1c. The absorbance band of 1,398 cm-1 belonged to B-O stretching vibration. The C=C stretching vibration of the benzene ring was at 1560 cm-1, and the peak of 1636 cm-1 was attributed to CO-NH stretching vibration, demonstrating 3-aminophenylboronic acid has been successfully grafted onto alginate. The grafting degree of phenylboronic acid groups was qualitatively estimated by 1H-NMR spectra of ALG-g-APBA with the equation proposed by Pettignano and co-workers [47]. The degree of substitution was calculated about 26% by the ratio between the integration areas of the phenylboronic acid unit and hydrogen in the alginate unit:
where IPBA is the integration areas of the phenylboronic acid and IALG are the integration areas of hydrogen in the alginate unit.

Fourier transform infrared (FTIR) spectrum of MSN (a), ALG (b), ALG-g-APBA (c), CS (d), and CS-g-FPBA (e)

1H-NMR spectrum of ALG (a), ALG-g-APBA (b), CS (c), and CS-g-FPBA (d)
The 1H-NMR spectrum of CS-g-FPBA is shown in Figure 2d. The chemical shifts at 2.8–4.0 were related to the resonance peak of protons of carbon skeleton in chitosan as well as the methyl peak of chitosan at 2.0 from incomplete acetylation. The peaks at δ=7.2–7.8 were assigned to protons of benzene ring in 3-carboxy-4-fluorophenylboronic acid, indicating that phenylboronic acid has been successfully grafted onto chitosan [48]. From the FTIR spectrum of CS-g-FPBA shown in Figure 1e, the characteristic peaks of CO-NH stretching vibration appeared at 1637 cm-1. The peaks at 1601 cm-1 were attributed to the stretching vibration of C=C skeleton on the aromatic ring. The peaks at 1371 cm-1 were assigned to substituted benzene absorption bands of bending vibration, revealing that 3-carboxy-4-fluorophenylboronic acid has been successfully grafted onto the glucosamine units of chitosan [49]. The degree of substitution was calculated about 15% by the ratio between the integration values of the phenylboronic acid unit and hydrogen in the chitosan unit as follows [50]:
where IPBA is the integration areas of the phenylboronic acid and ICS are the integration areas of hydrogen in the chitosan unit.
Fabrication and Characterization of Polyelectrolyte Complexes
The mesoporous silica (MSN) was considered a potential container for insulin due to the large surface area and tunable pore size. The morphology investigated by transmission electron microscope (TEM) showed smooth and spherical monodispersed particles with dendritic pores and an average diameter of approximately 165.4±2.7 nm (Figure 3a). The particle size measured by dynamic light scattering (DLS) was 187.2±2.7 nm, which was larger than TEM. It was because the hydrodynamic diameter measured in suspension was corresponded to hypothetical hard spheres dispersed in the same manner [51]. After wrapped with a polysaccharide layer of ALG-g-APBA and CS-g-FPBA, the surface became relatively rough (Figure 3b and c). The size of ALG-g-APBA@MSN and CS-g-FPBA@MSN nanoparticles increased to 274.0±9.5 nm and 257.1±4.4 nm, respectively, by DLS analysis, indicating that the polysaccharide has been successfully wrapped on MSN. As observed in Figure 3d, the PECs formed by ALG-g-APBA and CS-g-FPBA exhibited a crosslink nano-network. MSN still maintained dendritic pore structure after zooming in TEM images (Figure 3e). Furthermore, the zeta potential of MSN was −(25.5±1.0) mV, while increasing to +(16.8±1.0) mV after modification with APTES on the surface (MSN-NH2). Then, the zeta of ALG-g-APBA@MSN nanoparticles reversed to −(24.9±1.2) mV due to the successful package of negatively charged ALG-g-APBA on the positively charged MSN-NH2. The CS-g-FPBA@MSN nanoparticles are formed by coating the positively charged CS-g-FPBA on the negatively charged MSN, exhibiting a charge of +(20.0±0.3) mV (Figure 3f, Figure 4 Tables I, II).

Transmission electron microscope (TEM) images of MSN (a), ALG-g-APBA@MSN (b), CS-g-FPBA@MSN (c), PECs (d), magnified image of PECs (e), and zeta potential values of MSN, MSN-NH2, ALG-g-APBA@MSN, and CS-g-FPBA@MSN (F) (n=3).

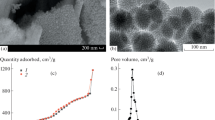
Nitrogen adsorption-desorption isotherms of MSN, ALG-g-APBA@MSN, CS-g-FPBA@MSN, and PECs (a); pore size distribution of MSN, ALG-g-APBA@MSN, CS-g-FPBA@MSN, and PECs (b).
Nitrogen adsorption-desorption analysis of MSN exhibited type-IV isotherm (Figure 5A), suggesting that MSN has an obvious mesoporous structure with an average pore size of 6.3 nm, pore volume of 1.47 cm3/g, and surface area of 750.4 m2/g, able to accommodate macromolecular drug as insulin [52]. When MSN nanoparticles were filled with insulin then, respectively, wrapped with ALG-g-APBA and CS-g-FPBA on the surface, the isotherm became obviously flat, and the pore sizes decreased to 2.8 nm and 2.5 nm. The pore volumes of ALG-g-APBA@MSN and CS-g-FPBA@MSN were 0.39 cm3/g and 0.28 cm3/g which were much less than MSN’s, indicating insulin has been loaded into the MSN interior. The surface area decreased to 218.5 m2/g and 127.1 m2/g, confirming that the surface of the MSN has been already coated by polymers. Moreover, the pore volume of PECs reduced to 0.15 cm3/g and the surface area to 76.7 m2/g owing to the cross-linked network on the surface of MSN. The PECs with a network possessed the capacity of preventing drug leakage.

The release profiles in vitro of PECs in different pH values and glucose concentrations at 37°C (n=3) (a); the switch “on-off” release profiles for PECs in 5-0-5-0 mg/mL of glucose (pH=7.4, 37°C) (n=3) (b); schematic representation for glucose responsiveness of PECs (c)
In Vitro Release Studies
The release performance in vitro of PECs was investigated under different glucose concentrations and pH conditions as shown in Figure 5a. In the gastric environment, the increasing protonation intensity of CS-g-FPBA leads to stronger electrostatic interaction with ALG-g-APBA, and the shrinking network can effectively prevent insulin from release or leakage. Thus, the cumulative release of insulin in the simulated gastric fluid of pH 1.2 was only 23.34±0.48% in total in 10 h and approximately 30.38±1.9% in 24 h. The glucose responsiveness profiles were explored with three glucose concentrations to simulate blood glucose levels in vivo, a control level (0 mg/mL), an euglycemic level (2 mg/mL), and a critical diabetic glucose level (5 mg/mL). The release rate of insulin in PBS buffer (pH=7.4) with glucose of 2 mg/mL was around 47.79±1.23% in 10 h and 66.21±4.34% in 24 h, which were obviously more than glucose-absence media of 33.09±2.58% and 47.44±2.38% but less than that with 5-mg/mL glucose of 78.07±2.60% and 90.92±3.11% at the same time points. It could be seen from the release curve that the release rate increases with the growth of glucose. 1,2-Diol moieties of diffused glucose competitively bind to PBA with PECs for breaking borate esters bond and swelling of network, resulting in glucose-dependent release behavior of insulin [35]. The network formed by electrostatic interaction and borate esters were in response to glucose and provided protection for insulin.
The blood glucose levels fluctuate for feeding, starving, and circadian rhythm of diabetic patients. It is essential to smartly regulate insulin release according to the blood glucose. To investigate the switch “on-off” of insulin release, the PECs were alternately immersed in 5 mg/mL of glucose solution and glucose-absence solution. As shown in Figure 5b, insulin released fast in glucose solution for the first 3 hours with the rate around 18.15±2.55%, whereas in glucose-absence solution only 4.01% for the next 3 hours. In four cycles of 5-0-5-0–mg/mL release profiles, insulin released a burst in glucose medium whereas a plateau in glucose-absence solution. It obviously demonstrated that the drug carriers played a role of a switch to control insulin release according to glucose concentrations. In the intestine, the network was swollen after 1,2-diol moieties of glucose competitively bond to PBA with CS-g-FPBA and ALG-g-APBA in high glucose condition, while shrinking following that CS-g-FPBA and ALG-g-APBA replaced glucose to recombine with the boronic acid group at glucose-absence medium (Figure 5c) [41, 53].
Cell Viability
In order to assess the effect of PECs on cell survival, Caco-2 cells were treated with blank CS-g-FPBA@MSN, ALG-g-APBA@MSN, and PECs at a series of concentrations (15.625, 31.25, 62.5, 125, 250, 500, 1000 μg/mL) for 24 hours and then measured by CCK-8 assay, which was widely used as a mitochondrion activity measurement to quantify the cell growth or cell death [54]. As shown in Figure 6, the cell viability for ALG-g-APBA@MSN, CS-g-FPBA@MSN, and PECs were all beyond 80% even at the highest concentration of 1000 μg/mL. It also could be seen that nanoparticles promoted the proliferation of Caco-2 cells at low concentrations. These results indicated that the nanoparticles had no toxicity on Caco-2 cells and could be used as an insulin delivery in vivo.

Cell viability of Caco-2 cells treated with blank ALG-g-APBA@MSN and CS-g-FPBA@MSN nanoparticles by CCK-8 assay after incubation for 24 h at 37°C (n=5)
In Vivo Release Studies
The hypoglycemic effects of different equations assessed on type I diabetic rats were described in Figure 7a. After oral administration in vivo, the PECs (INS) should be transported from the gastrointestinal tract which contains many obstacles (gastric acid, enzymes, mucus, and epithelial barrier). Subcutaneous injection could deliver insulin into blood capillaries directly between connective tissues. As a result, the bioactivity of subcutaneous injection is much higher than that of oral administration, and the dose of oral administration should be much higher than that of subcutaneous injection [55, 56]. Oral intake glucose solution (1 g/kg) post 30-min administration of insulin formulations was used to simulate the postprandial condition. As shown in Figure 7a, oral administration of glucose solution led to postprandial hyperglycemia, and the blood glucose levels of oral-administrated insulin (50 IU/kg) reduced to the initial level after 8 h while still maintained a high blood glucose state. The blood glucose levels of subcutaneous injection (5 IU/kg) decreased rapidly to the lowest value (14.19±3.48% of initial blood glucose) in 2 h and began to return to hyperglycemic level after 4 h, which was due to the short half-life of insulin [57]. PECs without PBA modification (50 IU/kg) acted as a control group that primordial alginate and chitosan were coated on MSN-NH2 and MSN nanoparticles, respectively. In this group, the blood glucose decreased to 56.93±10.25% of the initial level in 8 h and maintained a relatively constant decline tendency to 18 h. In comparison, PECs (INS) (50 IU/kg), triggered by high glucose level in the intestine after oral intake with glucose solution, exhibited a better hypoglycemic effect, which fell dramatically to 35.08±12.39% of the initial in 8 h but followed a slow and steady decline tendency to the lowest value (24.01±3.41%) till 14 h and then increased after 18 h. Owing to the modification of PBA, the PECs presented a rapid glucose responsiveness and self-regulation capacity which could maintain normal levels for nearly 12 h. Of note, the blood glucose of oral administration with PECs (INS) on normal rats exhibited almost no significant hypoglycemic effect but only a short and mild effect at 6 h. Therefore, PECs delivery system is capable of regulating release to achieve a steady and prolonged hypoglycemic effect [58].

Blood glucose levels of fasted STZ-induced type I diabetic rats after subcutaneous injection with insulin solution (5 IU/kg), oral PECs (50 IU/kg), oral PECs without PBA modification (50 IU/kg), oral insulin solution (50 IU/kg) in diabetic rats, and oral PECs (50 IU/kg) in normal rats (a); glucose solution (1 g/kg) was oral intake post 30-min administration of insulin formulations. Plasma insulin concentrations of subcutaneous injection with insulin solution (5 IU/kg), oral PECs (50 IU/kg), and oral PECs without PBA modification (50 IU/kg) in diabetic rats (b). Data points represent mean ± SD (n=5), significant difference between groups was analyzed with independent student’s t test: Both oral PECs without PBA modification (50 IU/kg) and oral insulin solution (50 IU/kg) were the control groups of two aspects, and oral PECs (50 IU/kg) was the experimental group. The t test was carried out twice because the experimental group should match each control group, respectively. Moreover, there was no need to adjust the significance probability because the two tests were not absolutely multiple or multi-index comparisons of sample groups from the same batch (α=0.05). Oral insulin solution (50 IU/kg) and oral PECs (50 IU/kg):*p<0.05, **p< 0.01, ***p < 0.001. Oral PECs without PBA modification (50 IU/kg) and oral PECs (50 IU/kg): #p<0.05
According to the pharmacokinetic curves in Figure 7B, the plasma insulin concentrations of diabetic rats in the subcutaneous injection group reached a maximum of 55.58±8.62 μIU/mL at 1 h and then dropped to the initial value after 3 h, indicating a fast elimination rate and short retention time of insulin in the body. In the group of PECs (INS) without PBA modification, the plasma insulin increased slowly to a max value of 10.87±5.08 μIU/mL at 10 h, whereas following a decrease to 3.43±3.07 μIU/mL to 18 h, indicating the insulin was completely released from PECs. By comparison, the plasma insulin concentrations in oral PECs (INS) group increased slightly in 3 h, after which it reached a peak value of 20.53±3.23 μIU/mL at 8 h. Subsequently, the plasma insulin concentration decreased comparatively stable from 10 h to 14 h because the network of PECs began to shrink to prevent insulin release at low glucose level. The plasma insulin was detected at a relatively low level (5.59±2.96 μIU/mL) at 14 h according with the slow decline tendency of blood glucose level. The relative bioavailability of PECs reached 11.40%, far more than that of insulin solution by oral administration (0.5%) [59] and approximately 1.5-fold higher than that of the PECs without PBA modification (8.53%). Thereby, PECs possessed a better bioavailability for the intelligent release regulation according with blood glucose variation.
Chitosan has the capacity for overcoming mucus and epithelial barrier due to good mucoadhesion to intestinal epithelium and promotion of paracellular transport through opening the tight junctions (TJs). It can prolong the retention time of insulin in the intestinal tract and enhance adsorption before being eliminated from the intestine [49, 60]. In addition, it is reported that calcium cation plays a vital role in the activity of digestive enzymes (such as trypsin) in the gastrointestinal tract. The carboxyl group on the alginate has a complexation effect on calcium cations, which can inhibit some digestive enzymes to improve the stability of the drug [61]. Overall, the orally administrated PECs with glucose responsiveness can be selected as a good candidate for a postprandial insulin delivery system.
CONCLUSION
Controlling the postprandial blood glucose level of diabetic patients is essential to achieve tight blood glucose control according to carbohydrate content before or after meals. This study developed an oral glucose-responsive delivery system for postprandial administration of insulin. The PECs were comprised of CS-g-FPBA@MSN nanoparticles and ALG-g-APBA@MSN nanoparticles through electrostatic interaction and borate esters, presenting a good pH stability and glucose responsiveness according to in vitro release studies. The self-regulation release profiles also manifested the PECs could switch “on” insulin release at hyperglycemic state and switch “off” release at euglycemic levels. Furthermore, in vivo studies on diabetic rats exhibited a significant hypoglycemic effect by self-regulating insulin release, and possessed a higher bioavailability. Therefore, the PECs have the potential to be used as a peroral glucose-triggered system to lessen the risk of glucose fluctuation and prolong release time.
References
Alberti G, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus. Provisional report of a WHO Consultation. Diabetic medicine : a journal of the British Diabetic Association. 1998;15:539–53.
Amer DA. 9. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes-2020 (vol 43, pg S98, 2020). Diabetes Care. 2020;43:1979.
Sonmez M, Ficai D, Ficai A, Alexandrescu L, Georgescu M, Trusca R, et al. Applications of mesoporous silica in biosensing and controlled release of insulin. Int J Pharm. 2018;549:179–200.
Agrawal AK, Gupta PN, Khanna A, Sharma RK, Chandrabanshi HK, Gupta N, et al. Development and characterization of in situ gel system for nasal insulin delivery. Die Pharmazie - An International Journal of Pharmaceutical Sciences. 2010;65(3):188–93.
Zhou X, Wu H, Long R, Wang S, Huang H. Xia Y, et al. Journal of Nanobiotechnology: Oral delivery of insulin with intelligent glucose-responsive switch for blood glucose regulation; 2020.
Singh S, Kushwah V, Agrawal AK, Jain S. Insulin- and quercetin-loaded liquid crystalline nanoparticles: implications on oral bioavailability, antidiabetic and antioxidant efficacy. Nanomedicine. 2018;13(5):521–37.
Agrawal AK, Kumar K, Swarnakar NK, Kushwah V, Jain S. “Liquid crystalline nanoparticles”: rationally designed vehicle to improve stability and therapeutic efficacy of insulin following oral administration. Mol Pharm. 2017;14(6):1874–82.
Agrawal AK, Harde H, Thanki K, Jain S. Improved stability and antidiabetic potential of insulin containing folic acid functionalized polymer stabilized multilayered liposomes following oral administration. Biomacromolecules. 2014;15(1):350–60.
Chen TT, Li SY, Zhu WT, Liang Z, Zeng QB. Self-assembly pH-sensitive chitosan/alginate coated polyelectrolyte complexes for oral delivery of insulin. J Microencapsul. 2019;36:96–107.
Wong CY, Al-Salami H. Dass CR. AAPS PharmSciTech: Lyophilisation improves bioactivity and stability of insulin-loaded polymeric-oligonucleotide nanoparticles for diabetes treatment; 2020.
Urimi D, Agrawal AK, Kushwah V, Jain S. Polyglutamic acid functionalization of chitosan nanoparticles enhances the therapeutic efficacy of insulin following oral administration. AAPS PharmSciTech. 2019;20(3):131.
Lang XQ, Wang T, Sun MJ, Chen XG, Liu Y. Advances and applications of chitosan-based nanomaterials as oral delivery carriers: a review. Int J Biol Macromol. 2020;154:433–45.
Yan C, Gu J, Lv Y, Shi W, Huang Z, Liao Y. 5-cholanic acid/glycol chitosan self-assembled nanoparticles (5-CHA/GC-NPs) for enhancing the absorption of FDs and insulin by rat intestinal membranes. AAPS PharmSciTech. 2019;20:30.
Agrawal AK, Urimi D, Harde H, Kushwah V, Jain S. Folate appended chitosan nanoparticles augment the stability, bioavailability and efficacy of insulin in diabetic rats following oral administration. RSC Adv. 2015;5(127):105179–93.
Abeer MM, Meka AK, Pujara N, Kumeria T, Strounina E, Nunes R, et al. Rationally designed dendritic silica nanoparticles for oral delivery of Exenatide. Pharmaceutics. 2019;11(8).
George M, Abraham TE. Polyionic hydrocolloids for the intestinal delivery of protein drugs: alginate and chitosan - a review. J Control Release. 2006;114:1–14.
Hua SB, Ma HZ, Li X, Yang HX, Wang A. pH-sensitive sodium alginate/poly(vinyl alcohol) hydrogel beads prepared by combined Ca2+ crosslinking and freeze-thawing cycles for controlled release of diclofenac sodium. Int J Biol Macromol. 2010;46:517–23.
Abeer MM, Rewatkar P, Qu Z, Talekar M, Kleitz F, Schmid R, et al. Silica nanoparticles: a promising platform for enhanced oral delivery of macromolecules. J Control Release. 2020;326:544–55.
Li LL, Liu TL, Fu CH, Tan LF, Meng XW, Liu HY. Biodistribution, excretion, and toxicity of mesoporous silica nanoparticles after oral administration depend on their shape. Nanomed-Nanotechnol Biol Med. 2015;11(8):1915–24.
Lamson NG, Berger A, Fein KC, Whitehead KA. Anionic nanoparticles enable the oral delivery of proteins by enhancing intestinal permeability. Nature Biomedical Engineering. 2020;4(1):84–96.
Qu Z, Wong KY, Moniruzzaman M, Begun J, Santos HA, Hasnain SZ, et al. One-Pot synthesis of pH-responsive Eudragit-mesoporous silica nanocomposites enable colonic delivery of glucocorticoids for the treatment of inflammatory bowel disease. Advanced therapeutics. 2021;4(2):2000165.
Raza A, Alavi SE, Sime FB, Han FY, Roberts JA, Popat A, et al. Microfluidic assembly of pomegranate-like hierarchical microspheres for efflux regulation in oral drug delivery. Acta Biomater. 2021;126:277–90.
Hou L, Zheng Y, Wang Y, Hu Y, Shi J, Liu Q, et al. Self-regulated carboxyphenylboronic acid-modified mesoporous silica nanoparticles with "touch switch" releasing property for insulin delivery. ACS Appl Mater Interfaces. 2018;10:21927–38.
Shen D, Yu H, Wang L, Khan A, Haq F, Chen X, et al. Recent progress in design and preparation of glucose-responsive insulin delivery systems. J Control Release. 2020;321:236–58.
Volpatti LR, Matranga MA, Cortinas AB, Delcassian D, Daniel KB, Langer R, et al. Glucose-responsive nanoparticles for rapid and extended self-regulated insulin Delivery. ACS Nano. 2020;14(1):488–97.
Yin R, Wang K, Du S, Chen L, Nie J, Zhang W. Design of genipin-crosslinked microgels from concanavalin A and glucosyloxyethyl acrylated chitosan for glucose-responsive insulin delivery. Carbohydr Polym. 2014;103(1):369–76.
Shi D, Ran M, Zhang L, Huang H, Li X, Chen M, et al. Fabrication of biobased polyelectrolyte capsules and their application for glucose-triggered insulin Delivery. ACS Appl Mater Interfaces. 2016;8:13688–97.
Yetisen AK, Jiang N, Fallahi A, Montelongo Y, Ruiz-Esparza GU, Tamayol A, et al. Glucose-sensitive hydrogel optical fibers functionalized with phenylboronic acid. Adv Mater. 2017;29:11.
Brooks WLA, Deng CC, Sumerlin BS. Structure–reactivity relationships in boronic acid–diol complexation. ACS Omega. 2018;3:17863–70.
Springsteen G, Wang B. A detailed examination of boronic acid–diol complexation. Tetrahedron. 2002;58:5291–300.
Matsumoto A, Ikeda S, Harada A, Kataoka K. Glucose-responsive polymer bearing a novel phenylborate derivative as a glucose-sensing moiety operating at physiological pH conditions. Biomacromolecules. 2003;4:1410–6.
Matsumoto A, Ishii T, Nishida J, Matsumoto H, Kataoka K, Miyahara Y. A synthetic approach toward a self-regulated insulin delivery system. Angewandte Chemie-International Edition. 2012;51:2124–8.
Shen DK, Yang JP, Li XM, Zhou L, Zhang RY, Li W, et al. Biphase stratification approach to three-dimensional dendritic biodegradable mesoporous silica Nanospheres. Nano Lett. 2014;14(2):923–32.
Juere E, Caillard R, Marko D, Del Favero G, Kleitz F. Smart protein-based formulation of dendritic mesoporous silica nanoparticles: toward oral delivery of insulin. Chem Eur J. 2020;26(23):5195–9.
Belbekhouche S, Charaabi S, Picton L, Le Cerf D, Carbonnier B. Glucose-sensitive polyelectrolyte microcapsules based on (alginate/chitosan) pair. Carbohydr Polym. 2018;184:144–53.
Mansour O, El Joukhar I, Belbekhouche S. H2O2-sensitive delivery microparticles based on the boronic acid chemistry: (phenylboronic-alginate derivative/dextran) system. React Funct Polym. 2019;145:104377.
Zhang D, Yu GH, Long Z, Yang GH, Wang B. Controllable layer-by-layer assembly of PVA and phenylboronic acid-derivatized chitosan. Carbohydr Polym. 2016;140:228–32.
Han CY, Huang HT, Dong Y, Sui XY, Jian BY, Zhu WQ. A comparative study of the use of mesoporous carbon and mesoporous silica as drug carriers for oral delivery of the water-insoluble drug carvedilol. Molecules. 2019;24(9).
Gu Z, Aimetti AA, Wang Q, Dang TT, Zhang YL, Veiseh O, et al. Injectable nano-network for glucose-mediated insulin delivery. ACS Nano. 2013;7(5):4194–201.
Zhao LL, Jin WL, Cruz JG, Marasini N, Khalil ZG, Capon RJ, et al. Development of polyelectrolyte complexes for the delivery of peptide-based subunit vaccines against group A streptococcus. Nanomaterials. 2020;10(5).
Hou L, Zheng YZ, Wang YC, Hu YR, Shi JJ, Liu Q, et al. Self-regulated carboxyphenylboronic acid-modified mesoporous silica nanoparticles with "touch switch" releasing property for insulin delivery. ACS Appl Mater Interfaces. 2018;10(26):21927–38.
Ji N, Hong Y, Gu ZB, Cheng L, Li ZF, Li CM. Chitosan coating of zein-carboxymethylated short-chain amylose nanocomposites improves oral bioavailability of insulin in vitro and in vivo. J Control Release. 2019;313:1–13.
Tian MP, Song RX, Wang T, Sun MJ, Liu Y, Chen XG. Inducing sustained release and improving oral bioavailability of curcumin via chitosan derivatives-coated liposomes. Int J Biol Macromol. 2018;120:702–10.
Li SY, Chen ZR, Wang J, Yan LB, Chen TT, Zeng QB. Fabrication and characterization of a novel semi-interpenetrating network hydrogel based on sodium carboxymethyl cellulose and poly(methacrylic acid) for oral insulin delivery. J Biomater Appl. 2020;35(1):3–14.
Chen XY, Wu W, Guo ZZ, Xin JY, Li JS. Controlled insulin release from glucose-sensitive self-assembled multilayer films based on 21-arm star polymer. Biomaterials. 2011;32(6):1759–66.
Jiao JQ, Fu JY, Wei YC, Zhao Z, Duan AJ, Xu CM, et al. Al-modified dendritic mesoporous silica nanospheres-supported NiMo catalysts for the hydrodesulfurization of dibenzothiophene: efficient accessibility of active sites and suitable metal-support interaction. J Catal. 2017;356:269–82.
Pettignano A, Grijalvo S, Haring M, Eritja R, Tanchoux N, Quignard F, et al. Boronic acid-modified alginate enables direct formation of injectable, self-healing and multistimuli-responsive hydrogels. Chem Commun. 2017;53(23):3350–3.
Shi DJ, Ran MS, Huang H, Zhang L, Li XJ, Chen MQ, et al. Preparation of glucose responsive polyelectrolyte capsules with shell crosslinking via the layer-by-layer technique and sustained release of insulin. Polym Chem. 2016;7(44):6779–88.
Smoum R, Rubinstein A, Srebnik M. Chitosan-pentaglycine-phenylboronic acid conjugate: a potential colon-specific platform for calcitonin. Bioconjug Chem. 2006;17(4):1000–7.
Li J, Wang PG, Zhang N, Yang Y, Zheng JB. Enhanced detection of saccharide using redox capacitor as an electrochemical indicator via a redox-cycling and its molecular logic behavior. Electrochim Acta. 2015;166:253–60.
Castangia I, Manca ML, Caddeo C, Maxia A, Murgia S, Pons R, et al. Faceted phospholipid vesicles tailored for the delivery of Santolina insularis essential oil to the skin. Colloid Surface B. 2015;132:185–93.
Tsuboi T, McMahon HT, Rutter GA. Mechanisms of dense core vesicle recapture following "kiss and run" ("cavicapture") exocytosis in insulin-secreting cells. J Biol Chem. 2004;279(45):47115–24.
Yu JC, Zhang YQ, Wang JQ, Wen D, Kahkoska AR, Buse JB, et al. Glucose-responsive oral insulin delivery for postprandial glycemic regulation. Nano Res. 2019;12(7):1539–45.
Leng D, Li Y, Zhu J, Liang RZ, Zhang CF, Zhou Y, et al. The antibiofilm activity and mechanism of nanosilver- and nanozinc-incorporated mesoporous calcium-silicate nanoparticles. Int J Nanomedicine. 2020;15:3921–36.
Li L, Jiang GH, Yu WJ, Liu DP, Chen H, Liu YK, et al. Preparation of chitosan-based multifunctional nanocarriers overcoming multiple barriers for oral delivery of insulin. Mat Sci Eng C-Mater. 2017;70:278–86.
Shan W, Zhu X, Liu M, Li L, Zhong JJ, Sun W, et al. Overcoming the diffusion barrier of mucus and absorption barrier of epithelium by self-assembled nanoparticles for oral delivery of insulin. ACS Nano. 2015;9(3):2345–56.
Ihlo CA, Lauritzen T, Sturis J, Skyggebjerg O, Christiansen JS, Laursen T. Pharmacokinetics and pharmacodynamics of different modes of insulin pump delivery. A randomized, controlled study comparing subcutaneous and intravenous administration of insulin aspart. Diabet Med. 2011;28(2):230–6.
Wang JQ, Yu J, Zhang YQ, Zhang XD, Kahkoska AR, Chen GJ, et al. Charge-switchable polymeric complex for glucose-responsive insulin delivery in mice and pigs. Science Advances. 2019;5(7).
Alibolandi M, Alabdollah F, Sadeghi F, Mohammadi M, Abnous K, Ramezani M, et al. Dextran-b-poly(lactide-co-glycolide) polymersome for oral delivery of insulin: in vitro and in vivo evaluation. J Control Release. 2016;227:58–70.
Sheng JY, Han LM, Qm J, Ru G, Li RX, Wu LH, et al. N-Trimethyl chitosan chloride-coated PLGA nanoparticles overcoming multiple barriers to oral insulin absorption. ACS Appl Mater Interfaces. 2015;7(28):15430–41.
Bravo-Osuna I, Millotti G, Vauthier C, Ponchel G. In vitro evaluation of calcium binding capacity of chitosan and thiolated chitosan poly(isobutyl cyanoacrylate) core-shell nanoparticles. Int J Pharm. 2007;338(1-2):284–90.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China under Grant [number 81373365] and Guangdong Science and Technology Project under Grant [number 2015A010105014].
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Qin, T., Yan, L., Wang, X. et al. Glucose-Responsive Polyelectrolyte Complexes Based on Dendritic Mesoporous Silica for Oral Insulin Delivery. AAPS PharmSciTech 22, 226 (2021). https://doi.org/10.1208/s12249-021-02088-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-021-02088-6