
Abstract
In a formulation, traces of peroxides in copovidone can impact the stability of drug substances that are prone to oxidation. The present study aimed to investigate the impact of peroxides in novel Plasdone™ S630 Ultra and compare it with regular Plasdone™ S630 on the oxidative degradation of quetiapine fumarate amorphous solid dispersions prepared via hot-melt extrusion technique. The miscibility of copovidones with drug was determined using the Hansen solubility parameter, and the results indicated a miscible drug–polymer system. Melt viscosity as a function of temperature was determined for the drug–polymer physical mixture to identify the suitable hot-melt extrusion processing temperature. The binary drug and polymer (30:70 weight ratio) amorphous solid dispersions were prepared at a processing temperature of 160°C. Differential scanning calorimetry and Fourier transform infrared spectroscopy studies of amorphous solid dispersions revealed the formation of a single-phase amorphous system with intermolecular hydrogen bonding between the drug and polymer. The milled extrudates were compressed into tablets by using extragranular components and evaluated for tabletability. Stability studies of the milled extrudates and tablet formulations were performed to monitor the oxidative degradation impurity (N-oxide). The N-oxide impurity levels in the quetiapine fumarate - Plasdone™ S630 Ultra milled extrudates and tablet formulations were reduced by 2- and 3-folds, respectively, compared to those in quetiapine fumarate - Plasdone™ S630. The reduced oxidative degradation and improved hot-melt extrusion processability of Plasdone™ S630 Ultra make it a better choice for oxidation-labile drugs over Plasdone™ S630 copovidone.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Trace levels of reactive impurities such as peroxides have been found in many pharmaceutical excipients, including povidones, copovidones, polyethylene glycol, and polysorbates [1]. The presence of peroxide impurities leads to the degradation of drug products containing oxygen-sensitive active pharmaceutical ingredients (APIs), resulting in decreased product performance, loss in potency, and formation of toxic degradation impurities [2]. The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use entails that the characterization and biological safety evaluation of degradation impurities are needed if the impurity profile is above the threshold levels based on daily dose [3]. Several approaches have been investigated for reducing the generation of peroxides from excipients, including chemical modification of the cross linker; use of enzymes and metals; and supercritical fluid extraction [2, 4, 5]. However, these approaches can limit the usage of excipients in a drug product because of the requirement of additional methods or additives for reducing peroxide concentration. Therefore, novel excipients such as Plasdone™ S630 Ultra (PS630U) copovidone, in which the initial peroxide levels as well as subsequent peroxide generation during storage can be controlled, were investigated as a choice of excipient for drug product stabilization against oxidative degradation. The molecular weight, particle size and distribution, flowability, and peroxide stability of the new PS630U copovidone were improved compared to those of regular Plasdone™ S630 (PS630) copovidone (Table I).
Copovidone is a synthetic random copolymer consisting of N-vinyl-2-pyrrolidone and vinyl acetate in 60:40 ratios. Copovidones have low glass transition temperature (Tg; 108–110°C) and are less hygroscopic [7]. Plasdone-grade copovidones are commonly used as a matrix polymer for amorphous solid dispersion (ASD) formulations to enhance the solubility and bioavailability of poorly soluble drugs [8]. However, the use of copovidones in ASDs is associated with stability concerns because of the presence of trace amounts of peroxides, which may accumulate upon storage and cause the degradation of APIs that are sensitive to oxygen. Copovidones may induce oxidative degradation via the following mechanisms: radical-initiated oxidation (autoxidation) and peroxide-mediated oxidation [9]. Drug substances that are sensitive to oxidation, especially APIs with piperazine and tertiary amine functional groups, are highly susceptible to interaction with copovidones via different mechanisms such as N-oxide formation (Fig. 1(a)) [10]. As per United States Pharmacopeia USP-35, the initial limit of peroxides in copovidones should not be more than 400 ppm (NMT 0.04%). Although the initial peroxide levels are within the limit, controlling the generation and growth of peroxides upon stability is necessary for drug product stabilization [11].

a Oxidation mechanism of quetiapine fumarate (QF) containing a piperazine group with peroxide impurities. b Potential known oxidative degradation impurities of QF
In the present study, quetiapine fumarate (QF), an oxidation-labile, atypical antipsychotic agent, was used as a model drug substance to determine the potential of PS630U and PS630 in the prevention of oxidative degradation when processed via hot-melt extrusion (HME). Quetiapine sulfoxide, quetiapine N-oxide derivative, and hydroxy quetiapine are the three oxidative degradation impurities of QF [12]. Previous studies have indicated that N-oxide derivative is one of the major oxidative impurities of QF [13]. Hence, in this study, the levels of quetiapine N-oxide (Quetiapine EP Impurity H) were measured as an oxidative impurity. The degradation pathway and structures of the oxidative impurities are shown in Fig. 1(b). This study aimed to investigate the potential of novel PS630U comparing it with that of PS630 in reducing the formation of oxidative impurities in tablets and milled extrudate formulations and evaluate the influence of the two copovidone grades on HME processability and QF ASD stability.
MATERIALS AND METHODS
Materials
Two types of copovidone grades PS630 and PS630U were supplied by Ashland, Inc. (Lexington, KY, USA). QF was obtained from RIA International LLC (East Hanover, NJ, USA). Avicel PH® 101 (microcrystalline cellulose) and Ac-Di-Sol® (croscarmellose sodium) were gift samples from FMC Biopolymer (Walnut Street, PA, USA); magnesium stearate and Aerosil® 200 (colloidal silica) were purchased from Alfa Aesar (Ward Hill, MA, USA). All other chemicals used were of analytical reagent grade.
Solubility Parameter Calculation
The miscibility of QF with Plasdone-grade copovidones was determined using the Hansen solubility parameters (δ) of the compounds by using group contributions from the chemical structures. The solubility parameter is a measure of the dispersion forces (δd), polarity forces (δp), and forces associated with hydrogen bonds (δh). The solubility parameter values were calculated using the Van Krevelen and Hoftyzer group contribution method by using the following equations [14]:
where δt is the total solubility parameter; δd, δp, and δh are the parameters associated with dispersive forces, polar forces, and hydrogen bonding, respectively; and Fdi, Fpi, and Ehi are the molar attraction constants due to dispersion, polar component, and hydrogen bonding energy, respectively. V is the molar volume. δ2 is commonly used to identify miscible drug–polymer combinations, and a system with similar δ2 is expected to be miscible [15].
Melt Rheology
The rheological properties of pure PS630 and PS630U polymers and binary physical mixture (PM) of QF-PS630 and QF-PS630U at a weight ratio of 30:70 were studied using an AR-G2 controlled-stress rotational rheometer (TA Instruments, New Castle, DE). A parallel plate geometry with plate diameter of 25 mm and gap distance of 1 mm was used. Disk specimens (slugs) of pure polymer and PM of QF–polymer were prepared by compressing approximately 1 g of each PM sample in a die by using a carver laboratory press. Dynamic temperature sweep was conducted across a temperature range of 130 to 180°C with a heating/cooling rate of 5°C/min to determine the effect of temperature on complex viscosity. Other experimental conditions used for dynamic temperature sweep were fixed strain of 0.2%, which was within the linear viscoelastic region to minimize microstructure destruction, and fixed angular frequency of 6.2 rad/s. The measurement temperature used for the study (130 to 180°C) was above the Tg and below the degradation temperature of the polymer. Viscoelastic properties such as G′ (elastic modulus), G″ (viscous modulus), and η* (complex viscosity) were recorded as a function of temperature.
HME Processing
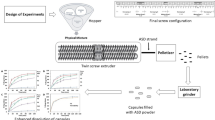
A binary PM of QF with PS630 and PS630U at a weight ratio of 30:70 was passed through a USP #25 mesh sieve, and the PM was mixed in a V-shell blender (Maxiblend, GlobePharma, NJ, USA) for 10 min at 20 rpm. HME was performed using a 16-mm Prism Euro Lab co-rotating twin-screw extruder (Thermo Fisher Scientific, Waltham, MA) consisting of 10 barrel zones. The PM was fed into the extruder by using a volumetric feeder (Brabender Technologies, Germany). The screw configuration consisted of 3 mixing zones. The kneading elements in the mixing zones were set at 90°, 60°, and 30° angle (mixing zone 1); 60° angle (mixing zone 2); and 90°and 60° angle (mixing zone 3) to facilitate adequate mechanical shear and allow dissolution of QF in the molten polymer. The extrusion process was performed at a screw speed of 100 rpm and feed rate of 10 g min−1. The barrel temperature for extrusion processing was identified on the basis of melt rheology analysis. During extrusion, the temperature of zone 1 was set at 50°C; for zones 2–9, temperatures were varied in the range of 140–160°C, and the temperature for zone 10 (near die region) was set at 140°C. The extrusion process parameters such as die pressure and torque values were monitored throughout the extrusion run. The obtained extrudates were cooled to ambient temperature and then milled using a laboratory-scale FitzMill (model L1A; Fitzpatrick, Perth Amboy, NJ, USA) by passing them through a 0.0200-in (equal to 500 μm) mesh screen by using an amplitude of 20 Hz at a speed of 1200 rpm. Milled extrudates were stored in an aluminum pouch until further processing.
Differential Scanning Calorimetry
Thermal analysis of QF, PS630, PS630U, and PM of QF–polymer and extruded samples was performed using Discovery differential scanning calorimetry (DSC) 25 (TA Instruments, New Castle, DE, USA), integrated with an RCS90 cooling device. In brief, approximately 5 mg of sample was weighed and placed in an aluminum pan, and then crimped with the lid on. The samples were equilibrated at 25°C for 1 min under nitrogen gas, followed by heating at 10°C per minute to 200°C under a nitrogen purge of 50 mL min−1. The Tg of the API, polymer, and milled extrudates was measured using the heat-cool-heat cycle, where the samples were heated to 200°C at 10°C mi−1 and then cooled to −20°C at a rate of 10°C min−1, followed by heating to 125°C at 2°C min−1. The thermograms were analyzed for Tg and melting temperature, to determine whether the crystalline drug had completely converted to the amorphous form after extrusion.
Fourier Transform Infrared Spectroscopy
The Fourier transform infrared (FTIR) spectra of the drug, polymers, PM, and extrudate formulations were obtained using an Agilent Cary 660 FTIR Spectrometer (Agilent Technologies, Santa Clara, CA, USA). A small amount of the sample was placed on the surface of a diamond crystal and pressed with a MIRacle high-pressure clamp. The spectra were recorded in the absorption mode from 600 to 4000 cm−1 wavenumber range with 16 samples/background scans and a resolution of 4 cm−1.
Tableting and Determination of Tensile Strength
Tablet formulations were developed using milled extrudates of QF-PS630, QF-S630U, and extragranular excipients such as fillers, superdisintegrants, lubricants, and glidants. The milled extrudates, microcrystalline cellulose (Avicel pH 101), and croscarmellose sodium (Ac-Di-Sol) were mixed in the V-shell blender for 10 min at 25 rpm. Next, colloidal silica and magnesium stearate were added and mixed in the V-shell blender for 3 min at 25 rpm. The final blend equivalent to 50 mg quetiapine base was compressed into a tablet (380 ± 10 mg) on a Piccola 10 station tablet compression machine equipped with a force feeder by using a 10-mm round concave tablet punching set at a compression force of 4.0 kN and turret speed of 12 rpm. Similarly, pure microcrystalline cellulose (MCC) and Plasdone™ copovidone tablets were also compressed. After compression, the tablets were characterized for weight, hardness, thickness, and diameter to determine the tensile strength (TS). The TS of the convex-faced tablets was calculated using the following formula [16, 17]:
where σx is the tensile strength, F is the breaking force, D is the tablet diameter, H is the tablet thickness, and W is the central cylinder thickness/tablet wall height.
High-Performance Liquid Chromatography (HPLC)
The oxidative impurity analysis of the samples was evaluated using high-performance liquid chromatography (HPLC; Waters Corp., Milford, MA, USA), where mobile phase (A) comprised 0.1% trifluoroacetic acid in DI water (v/v), and mobile phase (B) comprised methanol and acetonitrile at a ratio of 80:20 (v/v). The analytical column used was XBridge C18, 3.5 μm, and 100 × 4.6 mm, which was operated at 30°C with a flow rate of 0.5 mL min−1 and UV detection at 220 nm. The injection volume and run time were 15 μL and 60 min, respectively. For impurity analysis, sample solutions were prepared using the following procedure: The extrudates and tablet formulations equivalent to 20 mg of QF were accurately weighed into a 100-mL volumetric flask. Next, 75 mL of diluent (water:acetonitrile at 50:50 (v/v)) was added, and the resultant mixture was sonicated for 5 min, cooled to room temperature, diluted to final volume with the diluent, and then filtered. The filtered solution at a concentration of 206 μg mL−1 was transferred to an HPLC vial for analysis.
Dissolution Studies
The in vitro drug release studies of pure QF tablet and extrudate tablet formulations were performed at a dose equivalent to 50 mg quetiapine base (57.5 mg QF) in 900 mL of 0.05M phosphate buffer by using USP apparatus type I (SR8-plus™; Hanson), maintained at 37 ± 0.5°C with a speed of 100 rpm for 60 min (n = 3). Sample aliquots were collected at different time intervals by filtering through a 0.45-μm pore size PVDF membrane filter (Durapore®; Millipore Sigma, MA, USA) and analyzed for the amount of drug released by using HPLC.
Stability Study
Stability studies were conducted by placing milled extrudates and tablet formulations in high-density polyethylene bottles. The bottles were stored at 25°C/60% RH and 40°C/75% RH stability conditions. The milled extrudates were collected at 1 and 3 months (25°C/60% RH and 40°C/75% RH), and tablet formulations were collected at 1, 2, 3, and 6 months’ (40°C/75% RH) storage period. At each time point, samples were analyzed for the presence of oxidative impurities. The extrudate samples at 3-month 40°C/75% RH condition were analyzed using DSC to detect any recrystallization of QF in the ASD formulations.
RESULTS AND DISCUSSION
Solubility Parameter
The calculation of the Hansen solubility parameters for QF and copovidones depends on the intermolecular interactions, various types of cohesive and repulsive forces, and molecular volume of each component [18]. Depending on the functional groups present in the chemical structure of QF, the calculated solubility parameter (δ) for QF was 22.82 MPa1/2; the details are shown in Table II. The solubility parameter value for PS630 (26.40 MPa1/2) used in this study was obtained from the literature [18]. The functional groups present in both PS630U and PS630 copovidones are similar; hence, the solubility parameter values for both copovidone grades are comparable. Similar solubility parameter values of QF and Plasdone™ copovidones indicate miscibility. The difference in solubility parameters between the drug and polymer reflects the intermolecular interactions and miscibility of the drug within the polymer. A Δδ of < 7 MPa1/2 indicates miscibility, because of the balance between the energy of mixing from intermolecular and intramolecular interactions [19]. In contrast, Δδ of > 10 MPa1/2 indicates immiscibility [20] and might result in phase separation and/or recrystallization owing to the less prevalent intermolecular interactions. The difference in δ between QF and PS630 was found to be < 4 MPa1/2, suggesting the miscibility of QF within the polymer matrix. In this study, QF and Plasdone™ copovidones (PS630 and PS630U) had hydrogen bonding molar attraction constant values of 8.3 MPa1/2 and 11.86 MPa1/2, respectively. Because of their high molar attraction constant, intermolecular hydrogen bonding may play a main role in drug–polymer interactions [21].
δt QF = 22.82 Mpa1/2
Melt Rheology
In general, the rheological behavior can be measured as a function of temperature, time, frequency, and stress or strain amplitude. In this study, the melt rheology for pure PS630, pure PS630U, and PM of QF with both grades of copovidones was characterized by measuring the viscoelastic properties—η*, G′, and G″—as a function of temperature at constant strain and shear rate. Rheological analysis was performed in the linear viscoelastic region, where η*, G′, and G″ were measured by varying the stress or strain up to 100% while maintaining the temperature and frequency constant. Measuring the temperature-dependent melt viscosity is useful for predicting the HME process temperature. During temperature sweep, other parameters such as frequency and strain were held constant. The η*, G′, and G″ values of pure PS630, pure PS630U, and QF with PS630 and PS630U as a function of temperature are shown in Fig. 2. The η* refers to the resistance of the polymer or PM to flow. G′ is a measure of deformation, i.e., energy stored/recovered, and is related to molten polymer swelling. The loss of the rigid structure because of the applied temperature is referred to as G″, and it is a measure of energy lost or dissipated. The magnitude of crossover of G′ and G″ indicates the temperature at which the behavior of a material changes from elastic to viscous [22,23,24]. The plot of G′ and G″ as a function of temperature showed that these factors decrease as the temperature increases, and their curves intersect or reach close to each other at temperatures above 160°C and 180°C (Fig. 2(b, c)), respectively, for PM and pure Plasdone™ copovidones, suggesting a transition from a glassy to rubbery state [24].

a Complex viscosity profile as a function of temperature. b, c Plot of G′ and G″ versus temperature
The complex viscosity profiles of QF-PS630 and QF-PS630U PMs in the measured temperature range were less than those of pure PS630 and PS630U (Fig. 2(a)), indicating the plasticizing effect of QF. The extent of plasticizing may vary depending on the functional groups such as hydrogen bond donors and acceptors present in the API [25]. Previous studies suggested that a complex viscosity between 10,000 and 1000 Pa s should be ideal for optimal extrusion [26]. This range was established on the basis of the following considerations: (i) the polymer should be sufficiently less viscous to allow the dissolution of the drug; (ii) the plasticity of the polymer must be maintained so that the material can be extruded through the barrel and does not settle on the bottom of the barrel, thereby avoiding degradation; and (iii) extrudate strands need to exit from the die for successful extrusion [23, 27].
The η* values for pure PS630 and PS630U were approximately 10,000 Pa s at temperatures above 160°C (Fig. 2(a)) and decreased gradually with increasing temperature, and those of 30% QF with PS630 and PS630U were approximately 10,000 Pa s at above 135°C. Thus, the decreases in the complex viscosity of the PM below the melting temperature of QF suggested that the PM can be extruded well below the melting temperature of QF (174°C) owing to an increase in the separation of the copovidone polymer chains because of the plasticizing effect of QF and miscibility of QF in the polymer. These effects of QF may be attributed to the reduced complex viscosity of the PM compared to that of pure PS630 and PS630U [28]. The η* of PM decreased further when the temperature was increased to above 160°C, suggesting that QF might be completely miscible or might have dissolved into the polymer matrix.
Similarly, G′ and G″ for the PM decreased as the temperature increased (Fig. 2(b, c)); this magnitude of drop in G′ and G″ indicates a drop in elasticity and subsequent drop in complex viscosity. The plot of G′ and G″ as a function of temperature provides information regarding the amount of energy dissipated or lost in a sample. The higher value of G″ than that of G′ explains the viscoelastic or liquid state behavior of the sample as the temperature increases [23, 24]. The G″ > G′ in pure PS630, pure PS630U, and PM of QF-PS630 and QF-PS630U can be explained by the viscoelastic nature of the pure polymers and PM samples. These results suggest the plasticizing effect of QF, and the complex viscosity of PM samples was in the range of 10,000 to 1000 Pa s at a temperature range of 135–160°C. These observations aid in the selection of the extrusion processing temperature that does not generate high torque.
HME Processing
The melt viscosity results revealed that the lowest possible extrusion temperature for PM of QF copovidones was 135°C. In the case of pure PS630 and PS630U, extrusion was not possible at 140°C or below by using any screw configuration and feed rate because the torque exceeded the upper limit of the extruder [8]. However, PM at 30% drug load extruded successfully at 140°C with a processing torque value well below the instrument limit. This is because QF acted as a plasticizer for PS630 and PS630U, as confirmed by the melt rheology observations. The QF-PS630 PM (30:70) was extruded at 140, 150, and 160°C (zones 2–9) at a feed rate of 10 g min−1 and screw speed of 100 rpm (Table III). The copovidones were compared using the process conditions optimized with QF-PS630 PM extrusion for the extrusion of QF-PS630U PM.
The PS630 extrudates prepared at 140°C and 150°C appeared to be opaque with a rough surface, which could be attributed to the incomplete dissolution of crystalline QF. With increasing barrel temperature, torque decreased in the temperature range of 140–160°C (Table III), which reveals the shear-thinning behavior of the two Plasdone™ copovidone grades [29].
At 160°C, extrudates prepared with PS630 were brittle, transparent, and yellowish; for PS630U-containing formulations, extrudates were brittle, clear, and light yellowish, which could be attributed to the homogenous mixing. Crystalline QF dissolved into both copovidone grades. Furthermore, the yellowness decreased for PS630U extrudates, suggesting an improved performance and greater HME processability compared to those of the extrudates of PS630. The enhanced HME processability of PS630U could be attributed to the improvements in molecular weight, particle size distribution, and flowability compared to those of regular PS630. Traditionally, yellowness index analysis measures the sensitivity of a formulation to degradation and provides valuable information regarding the efficiency of the polymeric stabilizer. The greater the tendency of extrudates to turn yellow, the more is the sensitivity to degradation [30]. The images of PS630 and PS630U extrudates processed at different temperatures are shown in Fig. 3. During the extrusion, the processing torque and die melt pressure were in the range of 35–40% and 13–14 PSI, respectively, for formulations with both copovidone grades, suggesting that the selected process parameters were adequate and did not show excessive pressure generation inside the extruder. In the case of extrudates processed at 160°C, air bubbles were formed on the extrudates after the exit from the die. However, when the temperatures of zone 10 and die were decreased to 140°C, extrudates with little or no signs of air bubbles were produced.

Images of QF copovidone extrudates processed at different barrel temperatures
DSC
The thermal characteristics of QF, pure copovidones, and milled extrudates are shown in Fig. 4. The DSC thermogram of pure QF showed a sharp endothermic peak at 174°C (Fig. 4(a)) attributed to the melting of the crystalline drug [31]. The DSC thermogram of PM exhibited pronounced peak broadening and shifting of the QF melting point to a lower temperature value, which could be attributed to the gradual dissolution and/or miscibility of the drug into the polymer [32]. The extrudates of QF-PS630 prepared at a barrel temperature of 140°C and 150°C showed residual amount of crystalline QF, as evidenced by the small endothermic peak around 168°C (Fig. 4(a)). In contrast, extrudates prepared at 160°C showed no distinct endothermic peak of QF, indicating the complete transformation of crystalline QF into an amorphous form, suggesting the successful formation of ASDs via HME processing [33]. The heat-cool-heat cycle showed a Tg of 47°C, 108.7°C, and 110.6°C, respectively, for QF, PS630, and PS630U (Fig. 4(b)). The DSC analysis of the milled extrudates revealed the presence of a single Tg at 77.5°C and 79.8°C, respectively, for PS630 and PS630U extrudates, signifying the molecular-level miscibility of QF in the polymer. For an ASD, a distinctive single Tg has been considered an indication of molecular-level dispersion or dissolution of a drug in the polymeric system, which is critical for the physical stability of ASDs against crystallization [34, 35]. For both copovidone grades, the Tg of the formulation did not vary considerably and showed an identical single Tg, confirming the similarity in homogeneous and molecular-level mixing of the drug and polymer.

a DSC thermograms of pure QF, PS630, PS630U, PM, and extrudates processed at 140°C, 150°C, and 160°C. b Glass transition temperatures of QF, PS630U, PS630, and the extrudates of QF-PS630 and QF-PS630U
FTIR
The FTIR spectra were used to characterize and understand the possible hydrogen bonding and drug–polymer interactions of the QF-PS630 and QF-PS630U ASDs. The FTIR spectra of pure QF, pure PS630, pure PS630U, PM, and extruded formulations are shown in Fig. 5. The FTIR spectra of QF showed characteristic bands of C=O at 1599 cm−1 and the O–H stretching vibrations at 3250–3500 cm−1. The characteristic peaks at 2887 and 2945 cm−1 corresponded to the –C–H stretching vibrations. The peaks at 3100 cm−1 were attributed to aromatic =C–H stretching from the thiazepine ring. The peaks at 1260 and 1305 cm−1 corresponded to C–O–C asymmetrical stretching and C–N stretching, respectively [36]. The Plasdone™ copovidones showed characteristic bands of C=O stretch of vinyl acetate and amide carbonyl at 1731 and 1670 cm−1, respectively. The PM spectra showed frequency bands corresponding to individual components, suggesting the absence of interaction between the drug and polymer in the PM sample [37]. In the FTIR spectrum of milled extrudates, the characteristic C=O stretch of the amide carbonyl shifted from 1670 to 1656 cm−1 (QF-PS630U) and 1661 cm−1 (QF-PS630), and a new peak was observed at 1598 cm−1, indicating intermolecular hydrogen bonding between the drug and polymer. Hydrogen bonding between QF and Plasdone™ copovidones can disrupt the polymer chain arrangement, which results in the plasticization effect of QF [28].

The FTIR spectra of a pure QF; b PS630; c PS630U; d PM of QF-PS630; e PM of QF-PS630U; and f, g extrudates of QF-PS630 and QF-PS630U respectively
The literature states that the vinyl acetate C=O carbonyl group of copovidones is a weak hydrogen bond acceptor, and the hydrogen bond–donating functional groups in drug molecules are likely involved in intermolecular interactions and restrict crystallization. Furthermore, the strong intermolecular interactions between the drug and polymer might account for the improved ability of the polymers to inhibit nucleation and crystallization [38, 39]. In addition, the reduced intensities of the characteristic bands of QF in the formulation might be attributed to the molecular-level distribution of QF in the copovidone polymers.
Tableting and Tensile Strength Measurement
From the downstream processing perspective, a substantial amount of extragranular excipients was required to improve the flowability, compressibility, and disintegration properties of the milled extrudates. Initially, milled extrudates at a level of 60–70% with 40–30% of extragranular excipients were evaluated for their compression feasibility. The tableting blend at these levels of milled extrudates exhibited poor compression characteristics, which could be attributed to the denser, less internal voids; brittleness of the extruded filaments; and lower porosity of the tablet blend. During compression, the force required to compress a tablet blend containing 60% and 70% level of milled extrudates was 15 and 20 kN, respectively. Furthermore, decreasing the milled extrudate level to 50% and increasing the MCC level to 40% resulted in a decrease in compression force to 4.2 kN, suggesting increased porosity of the tablet blend by increasing the MCC level. The high intraparticle porosity and greater surface area characteristics of MCC could promote the tableting of the milled extrudates [40]. Tabletability is described as the magnitude of a powder blend to produce tablets of specific tensile strength under the influence of compression force [41]. The compressed tablets were characterized for breaking force (KP), diameter (mm), and thickness (mm) in order to calculate the tensile strength. The tensile strength of the milled extrudates, pure PS630, PS630U, and MCC, is shown in Fig. 6, and the composition of the tablets selected for tensile strength measurement is provided in Table IV.

Tensile strength of tablets prepared from QF-PS630 and QF-PS630U milled extrudates, pure PS630, PS630U, and MCC
The tablets of pure MCC and copovidones (PS630, PS630U) exhibited high TS, which could be attributed to the inter-particulate bonding or inherent adhesion properties of the material within a tablet under the applied compression force [42, 43]. In contrast, a tableting blend containing milled extrudates showed lower TS owing to the increase in particle density and loss of voids caused by heat and shear stress during HME [44]. This suggests that melt extrusion reduces the ability of milled extrudates to form strong bonds during compression. These observations were in accordance with those of previous studies. Iyer et al. [45] evaluated the impact of HME on the TS of tablets containing milled extrudates of copovidone and compared it with that of unprocessed or pure copovidone tablets. The results indicated that the TS of copovidone tablets decreased upon HME compared to that of the tablets of pure copovidone owing to the brittle nature of the extrudates formed upon melt extrusion, suggesting the densification and poor bonding properties of milled extrudates.
Dissolution Studies
The drug release of pure QF tablets and QF-PS630 and QF-PS630U tablet formulations was performed in 0.05M phosphate buffer as a dissolution medium (Fig. 7). The pure QF and tablet formulations with both grade copovidones showed 100% drug release within 15 min. This rapid dissolution rate could be attributed to the high solubility of QF in the dissolution medium. During the initial time points, the dissolution rate was relatively slow for tablet formulations, which can be described by polymer hydration and formation of a gel layer on the surface of the tablets [46].

The dissolution profile of QF-PS630U, QF-PS630, and pure QF tablets
QF exhibits a pH-dependent solubility profile with a high solubility in the pH range of 1.2–6.8. The reported solubility was found to be >9.2 mg mL−1 in 50 mM phosphate buffer [47]. Because of the high solubility in the studied dissolution media, no limitations of supersaturation and precipitation phenomenon of QF were observed during the dissolution study, and copovidone grades had no significant influence on the dissolution rate.
The saturation solubility (Cs) to the dose (Cd) ratio of QF in the dissolution media was notable. The calculated Cs and Cd of QF in phosphate buffer were found to be 9.2 and 0.064 mg mL−1, respectively. These results indicated that the dissolution rate may not be a rate-limiting step for QF.
Stability Study
The milled extrudates and tablet formulations of QF-PS630 and QF-PS630U subjected to long-term (25°C/60% RH) and accelerated (40°C/75% RH) stability conditions were analyzed to determine the oxidative degradation profile. The oxidative impurities of the milled extrudates and tablet formulations of QF-PS630 and QF-PS630U during the stability study are shown in Fig. 8.

Oxidative degradation profile of a, b milled extrudates and c tablet formulations
The PS630U milled extrudates had the lowest percent oxidative impurity with 0.19% and 0.42% at 3 months of 25°C/60% RH (Fig. 8(a)) and 40°C/75% RH (Fig. 8(b)) conditions, respectively. In contrast, the extrudates of PS630 showed relatively high percentage of oxidative impurities (0.42% and 0.95%, respectively) at 25°C/60% RH and 40°C/75% RH stability conditions, respectively.
For QF-PS630 tablet formulations (Fig. 8(c)), the 6-month stability at 40°C/75% RH condition showed an oxidative impurity of 0.39%. In contrast, tablets prepared with QF-PS630U showed 0.13% oxidative impurity, which is 3-fold less than that of tablets developed with regular PS630-grade copovidones. The increase in oxidative impurity at accelerated stability conditions (40°C/75% RH) suggests the temperature-dependent oxidative degradation of QF in the ASD formulations. For PS630U formulations, the lower levels of oxidative impurities could be attributed to the low initial level of peroxides present in PS630U and the generation of reactive impurities during storage, as revealed by stability studies compared to regular PS630-grade copovidone formulations. Peroxides are reactive impurities present in various copovidone-grade polymers that can directly react with the piperazine moiety of QF, initiate radical chain reactions, and induce the formation of oxidative degradation impurities [48].
Milled extrudates of both PS630 and PS630U showed variable oxidative degradation profiles with 2.2- and 2.4-fold less oxidative impurities in PS630U than in PS630 at 25°C/60% RH and 40°C/75% RH conditions, respectively. The oxidative impurity levels clearly increased for both the milled extrudates and tablet formulations. In addition, the initial level of oxidative impurities for QF-PS630 milled extrudates was found to be 0.15%. In the case of QF-PS630U milled extrudates, no oxidative impurities (below the limit of detection) were present, suggesting the existence of very low levels of peroxides in PS630U-grade copovidones.
Furthermore, the mechanism responsible for the oxidative degradation of QF could be either free radical chain mechanism or direct tertiary amine oxidation due to peroxide impurities present in the Plasdone™ copovidone polymers [11, 49]. Peroxides can be either organoperoxides (ROOR) or hydroperoxides (ROOH). The free radical chain mechanism for peroxide generation involves the cleavage of the bonds, followed by the addition of oxygen next to a hetero atom, which leads to the formation of peroxy free radicals [2].
While designing new formulations comprising oxygen-sensitive APIs, considering the initial peroxide levels and potential growth of reactive impurities of copovidones upon stability is necessary. The oxidative impurity data suggested that PS630U had better stability and prevented peroxide-induced oxidative degradation under stability conditions compared to regular PS630-grade copovidones.
The physical stability was evaluated by characterizing the extrudate formulations by using DSC after storage for 3 months under 40°C/75% RH stability condition. The melting thermograms of the stability samples are shown in Fig. 9.

DSC thermograms of QF-PS630 a and QF-PS630U b extrudates after 3 months of storage at 40°C/75% RH
DSC thermograms of stability samples showed that QF with PS630 and PS630U remained amorphous in the formulations with both copovidone grades and the slight endothermic peak observed at a temperature of approximately 100°C could be attributed to the evaporation of moisture during DSC analysis. The DSC results of the stability samples confirmed the homogeneity, and no phase separation of the amorphous drug was noted, as evidenced by the absence of the melting peak of QF, suggesting that HME formulations were physically stable during the stability study.
CONCLUSION
The impact of peroxide levels of PS630 and PS630U on oxidative degradation was successfully studied using the HME technique. Melt viscosity results provided insights into the selection of the HME processing temperature of both copovidone grades and exhibited similar complex viscosity profiles as a function of temperature. The extrudates of PS630U were transparent and light yellow in color, and PS630 extrudates appeared as dark yellow. These observations could be attributed to the superior HME processability of PS630U compared to that of regular PS630 copovidone. Thermal characterization and tabletability of both PS630U and PS630 formulations showed comparable Tg and TS profiles, respectively, indicating similar physicochemical characteristics between PS630 and PS630U copovidones. The stability study of the milled extrudates and tablet formulations confirmed that QF is highly sensitive to trace amounts of peroxides in copovidones, which caused the oxidation of QF to generate N-oxide impurities. The low initial peroxide levels, reduced growth, and peroxide generation in PS630U, as evidenced by low oxidative impurities after 6M stability studies, suggested that PS630U is relatively stable and showed less peroxide generation at ambient and accelerated stability conditions compared to regular PS630. Furthermore, the outcome from this study revealed the applicability of the novel Plasdone™ 630 Ultra for improved drug product stability against oxidative degradation in ASDs developed using the HME technique.
References
Wasylaschuk WR, Harmon PA, Wagner G, Harman AB, Templeton AC, Xu H, et al. Evaluation of hydroperoxides in common pharmaceutical excipients. J Pharm Sci. 2007;96:106–16.
Narang AS, Rao VM, Desai DS. Effect of antioxidants and silicates on peroxides in povidone. JPharmSci Elsevier. 2012;101:127–39.
Guideline IHT. Q3B (R2) Impurities in new drug products (2006). The international conference on harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH).
Hovorka SW, Schöneich C. Oxidative degradation of pharmaceuticals: theory, mechanisms and inhibition. J Pharm Sci. 2001;90:253–69.
Tallon MA, Malawer EG, Machnicki NI, Brush PJ, Wu CS, Cullen JP. The effect of crosslinker structure upon the rate of hydroperoxide formation in dried, crosslinked poly (vinylpyrrolidone). Journal of applied polymer science. Wiley Online Library. 2008;107:2776–85.
Ashland | pharmaceutical project report on PlasdoneTM S630 Ultra and PlasdoneTM S630.
Moroni A. A novel copovidone binder for dry granulation and direct-compression tableting. Pharm Technol ASTER PUBL CORP. 2001;25:8–13.
Zecevic DE, Evans RC, Paulsen K, Wagner KG. From benchtop to pilot scale–experimental study and computational assessment of a hot-melt extrusion scale-up of a solid dispersion of dipyridamole and copovidone. International journal of pharmaceutics. Elsevier. 2018;537:132–9.
Baertschi SW, Alsante KM, Reed RA. Pharmaceutical stress testing: predicting drug degradation, Second Edition. CRC Press; 2016.
Koo OMY. Pharmaceutical excipients: properties, functionality, and applications in research and industry. John Wiley & Sons; 2016.
Prachi S, Komal C, Priti MJ. Influence of peroxide impurities in povidone on the stability of selected β-blockers with the help of HPLC. AAPS PharmSciTech. 2017;18:2410–7.
de Diego M, Campos C, Correa D, Mennickent S, Godoy R, Vergara C. Degradation studies of quetiapine fumarate by liquid chromatography–diode array detection and tandem mass spectrometry methods. In: Biomedical Chromatography, vol. 33: Wiley Online Library; 2019. p. e4655.
Trivedi RK, Patel MC. Development and validation of a stability indicating RP-UPLC method for determination of quetiapine in pharmaceutical dosage form. Sci Pharm. 2011;79:97–111.
Just S, Sievert F, Thommes M, Breitkreutz J. Improved group contribution parameter set for the application of solubility parameters to melt extrusion. European Journal of Pharmaceutics and Biopharmaceutics. Elsevier. 2013;85:1191–9.
Verma S, Rudraraju VS. A systematic approach to design and prepare solid dispersions of poorly water-soluble drug. AAPS PharmSciTech. 2014;15:641–57.
Pitt KG, Newton JM, Richardson R, Stanley P. The material tensile strength of convex-faced aspirin tablets. Journal of pharmacy and pharmacology. Wiley Online Library; 1989;41:289–292.
Shang C, Sinka IC, Jayaraman B, Pan J. Break force and tensile strength relationships for curved faced tablets subject to diametrical compression. Int J Pharm. 2013;442:57–64.
Hurley D, Carter D, Ng LYF, Davis M, Walker GM, Lyons JG, et al. An investigation of the inter-molecular interaction, solid-state properties and dissolution properties of mixed copovidone hot-melt extruded solid dispersions. J Drug Delivery Sci Technol Elsevier. 2019;53:101132.
Forster A, Hempenstall J, Tucker I, Rades T. Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis. International journal of pharmaceutics. Elsevier. 2001;226:147–61.
Ghebremeskel AN, Vemavarapu C, Lodaya M. Use of surfactants as plasticizers in preparing solid dispersions of poorly soluble API: selection of polymer–surfactant combinations using solubility parameters and testing the processability. International journal of pharmaceutics. Elsevier. 2007;328:119–29.
Maniruzzaman M, Snowden MJ, Bradely MS, Douroumis D. Studies of intermolecular interactions in solid dispersions using advanced surface chemical analysis. RSC advances. Royal Soc Chem. 2015;5:74212–9.
Lu J, Obara S, Ioannidis N, Suwardie J, Gogos C, Kikuchi S. Understanding the processing window of hypromellose acetate succinate for hot-melt extrusion, part I: polymer characterization and hot-melt extrusion. Advances in polymer technology. Wiley Online Library. 2018;37:154–66.
Gupta SS, Solanki N, Serajuddin ATM. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion, IV: AffinisolTM HPMC HME polymers. AAPS PharmSciTech. 2016;17:148–57.
Gupta SS, Meena A, Parikh T, Serajuddin AT. Investigation of thermal and viscoelastic properties of polymers relevant to hot melt extrusion-I: polyvinylpyrrolidone and related polymers. J Excipients Food Chem Int Pharm Excipients Council Am. 2016;5:1001.
Yang F, Su Y, Small J, Huang C, Martin GE, Farrington AM, et al. Probing the molecular-level interactions in an active pharmaceutical ingredient (API) - polymer dispersion and the resulting impact on drug product formulation. Pharm Res. 2020;37:94.
Solanki N, Gupta SS, Serajuddin AT. Rheological analysis of itraconazole-polymer mixtures to determine optimal melt extrusion temperature for development of amorphous solid dispersion. European J Pharm Sci Elsevier. 2018;111:482–91.
Butreddy A, Bandari S, Repka MA. Quality-by-design in hot melt extrusion based amorphous solid dispersions: an industrial perspective on product development. Eur J Pharm Sci. 2021;158:105655.
Dudhedia MS, Agrawal AM. Rheological study of copovidone and solid dispersion blend used for hot melt extrusion. J Appl Polym Sci. Wiley Online Library. 2016:133.
Ma X, Huang S, Lowinger MB, Liu X, Lu X, Su Y, et al. Influence of mechanical and thermal energy on nifedipine amorphous solid dispersions prepared by hot melt extrusion: preparation and physical stability. Int J Pharm Elsevier. 2019;561:324–34.
Sarode AL, Obara S, Tanno FK, Sandhu H, Iyer R, Shah N. Stability assessment of hypromellose acetate succinate (HPMCAS) NF for application in hot melt extrusion (HME). Carbohydr Polym. 2014;101:146–53.
Li Y, Lu M, Wu C. PVP VA64 as a novel release-modifier for sustained-release mini-matrices prepared via hot melt extrusion. Drug delivery and translational research. Springer. 2018;8:1670–8.
Li M, Gogos CG, Ioannidis N. Improving the API dissolution rate during pharmaceutical hot-melt extrusion I: effect of the API particle size, and the co-rotating, twin-screw extruder screw configuration on the API dissolution rate. International journal of pharmaceutics. Elsevier. 2015;478:103–12.
Butreddy A, Sarabu S, Dumpa N, Bandari S, Repka MA. Extended release pellets prepared by hot melt extrusion technique for abuse deterrent potential: category-1 in-vitro evaluation. Int J Pharm. 2020;119624.
Qian F, Huang J, Zhu Q, Haddadin R, Gawel J, Garmise R, et al. Is a distinctive single Tg a reliable indicator for the homogeneity of amorphous solid dispersion? International journal of pharmaceutics. Elsevier. 2010;395:232–5.
Wani RJ, Sharma P, Zhong HA, Chauhan H. Preparation and characterization of griseofulvin solid dispersions. Assay Drug Dev Technol. 2020;18:109–18.
Gohel MC, Patel TM. Compatibility study of quetiapine fumarate with widely used sustained release excipients. J Therm Anal Calorim. 2013;111:2103–8.
Butreddy A, Sarabu S, Bandari S, Dumpa N, Zhang F, Repka MA. Polymer-assisted aripiprazole–adipic acid cocrystals produced by hot melt extrusion techniques. Crystal Growth & Design. Am Chem Soc. 2020;20:4335–45.
Frank DS, Matzger AJ. Probing the interplay between amorphous solid dispersion stability and polymer functionality. Molecular Pharm ACS Publ. 2018;15:2714–20.
Yuan X, Xiang T-X, Anderson BD, Munson EJ. Hydrogen bonding interactions in amorphous indomethacin and its amorphous solid dispersions with poly (vinylpyrrolidone) and poly (vinylpyrrolidone-co-vinyl acetate) studied using 13C solid-state NMR. Molecular Pharm ACS Publ. 2015;12:4518–28.
Thoorens G, Krier F, Leclercq B, Carlin B, Evrard B. Microcrystalline cellulose, a direct compression binder in a quality by design environment—a review. Int J Pharm. 2014;473:64–72.
Tye CK, Sun CC, Amidon GE. Evaluation of the effects of tableting speed on the relationships between compaction pressure, tablet tensile strength, and tablet solid fraction. Journal of pharmaceutical sciences. Elsevier. 2005;94:465–72.
Abdullah AHD, Putri OD, Fikriyyah AK, Nissa RC, Intadiana S. Effect of microcrystalline cellulose on characteristics of cassava starch-based bioplastic. Polymer-Plastics Technology and Materials. Taylor & Francis; 2020;59:1250–1258.
Patel S, Kou X, Hou HH, Huang YB, Strong JC, Zhang GG, et al. Mechanical properties and tableting behavior of amorphous solid dispersions. J Pharm Sci Elsevier. 2017;106:217–23.
Agrawal A, Dudhedia M, Deng W, Shepard K, Zhong L, Povilaitis E, et al. Development of tablet formulation of amorphous solid dispersions prepared by hot melt extrusion using quality by design approach. AAPS PharmSciTech. 2016;17:214–32.
Iyer R, Hegde S, Zhang Y-E, Dinunzio J, Singhal D, Malick A, et al. The impact of hot melt extrusion and spray drying on mechanical properties and tableting indices of materials used in pharmaceutical development. Journal of pharmaceutical sciences. Elsevier. 2013;102:3604–13.
Maincent J, Williams RO. Sustained-release amorphous solid dispersions. Drug Deliv and Transl Res. 2018;8:1714–25.
Hamed R, AlJanabi R, Sunoqrot S, Abbas A. The effect of pH, buffer capacity and ionic strength on quetiapine fumarate release from matrix tablets prepared using two different polymeric blends. Drug Development and Industrial Pharmacy. Taylor & Francis; 2017;43:1330–1342.
Júlio TA, Zâmara IF, Garcia JS, Trevisan MG Compatibility and stability of valsartan in a solid pharmaceutical formulation. Brazilian Journal of Pharmaceutical Sciences. Faculdade de Ciências Farmacêuticas da Universidade de São Paulo; 2013;49:645–651.
Wu Y, Levons J, Narang AS, Raghavan K, Rao VM. Reactive impurities in excipients: profiling, identification and mitigation of drug–excipient incompatibility. AAPS PharmSciTech. 2011;12:1248–63.
Funding
This work was partially supported by the National Institute of General Medical Sciences (NIGMS), a component of the National Institutes of Health (NIH) as one of its Centers of Biomedical Research Excellence (COBRE), under Grant Number P30GM122733-01A1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editors: Harsh Chauhan and Robert (Bill) Williams III
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Butreddy, A., Sarabu, S., Bandari, S. et al. Influence of Plasdone™ S630 Ultra—an Improved Copovidone on the Processability and Oxidative Degradation of Quetiapine Fumarate Amorphous Solid Dispersions Prepared via Hot-Melt Extrusion Technique. AAPS PharmSciTech 22, 196 (2021). https://doi.org/10.1208/s12249-021-02069-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-021-02069-9