
Abstract
Lipid-based drug delivery systems (LbDDS), such as self-nanoemulsifying drug delivery systems (SNEDDS), constitute a prominent formulation approach for enhancing the aqueous solubility and oral bioavailability of poorly water-soluble compounds. Utilization of biorefinery wastes, such as oil from rice bran, may prove advantageous to both improving drug solubilization and absorption and to achieving sustainable agri-food waste valorization. Here, we assessed the effect of four SNEDDS compositions differing in the oil (rice bran oil and corn oil) and surfactant type (Kolliphor RH40 and EL) on the oral bioavailability of fenofibrate, a BCS class II compound. Prior to the in vivo oral administration of the SNEDDS in rats, drug solubilization was tested in vitro using the static digestion model, followed by the ex vivo permeability study of the predigested SNEDDS using the non-everted gut sac model. No significant variation was observed in the solubilization capacity within the different SNEDDS formulations. On the other hand, the ex vivo permeability data of the predigested SNEDDS correlated well with the in vivo bioavailability data designating the superiority of rice bran oil with Kolliphor EL as the surfactant, to enhance the oral absorption of fenofibrate. Results indicated that valorization of agro-industrial waste such as rice bran oil may prove useful in enhancing the oral performance of LbDDS in the case of fenofibrate, while at the same time maximizing the use of agricultural by-products via the creation of new sustainable value chains in the pharmaceutical field.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Lipid-based drug delivery systems (LbDDS), such as self-nanoemulsifying drug delivery systems (SNEDDS), have been utilized as a prominent approach for the oral formulation development of lipophilic drug compounds with poor aqueous solubility (BCS class II drugs) (1,2). Since the intestinal absorption is a dissolution rate-limiting process, LbDDS have the potential to improve bioavailability by enhancing drug solubilization in the gastrointestinal fluids, prolonging residence time in the gastrointestinal tract (GIT), stimulating biliary and pancreatic secretions and lymphatic transport, and by increasing intestinal permeability (3). However, oral drug absorption is a dynamic process highly dependent on a combination of complex events occurring simultaneously (4) that lie on the interface of the evolution of the solubilization capacity of the colloidal dispersions that are formed during digestion of the LbDDS and intestinal drug permeability (5).
The introduction of in vitro models (6,7) that are able to simulate digestion independently of the use of costly equipment as an alternative to pH-stat lipolysis models, has facilitated a more simplified and efficient approach to LbDDS screening. At the same time, combined digestion-permeation models, either in a simultaneous or a consecutive mode, have been evaluated for their predictive potency in pursuit of a more physiologically relevant procedure to simulate drug absorption (8). Combined models using Caco-2 cell monolayers (9), intestinal rat tissue (10), or artificial membrane (11) as the permeability barrier were found to adequately predict and rank the oral performance of LbDDS. Further research is yet required in order to verify these models and equally important to minimize their complexity, cost, and low throughput.
Natural oils of plant origin or purified fractions of them are common excipients in self-emulsifying drug delivery systems (SEDDS) given their generally recognized as safe (GRAS) status and their use as food constituents worldwide (12). Lately, efforts are focusing on exploring non-conventional sources for oil extraction, including agro-industrial by-products (13). This strategy not only contributes to the circular economy but also yields the conversion of what would have been considered as waste into highly valuable products (14). Rice bran oil (RBO) is extracted from a by-product of the rice milling process (15). The annual global rice production exceeds 700 million metric tons leaving an immense potential for RBO production of approximately 3–4 million metric tons (16). RBO has a fatty acid composition of 43% monounsaturated and 35% polyunsaturated fatty acids and is well known for its nutritional value and health benefits, among which its hypocholesterolemic, anti-diabetic, antioxidant, anti-inflammatory and anti-cancer activities (17,18,19,20). Taking into account the health prospects of RBO and that valorization of by-products from natural sources starts gaining significant impetus, we evaluated RBO as a lipidic carrier in SNEDDS formulations to enhance the oral bioavailability of fenofibrate, a BCS class II active compound. So far, RBO has only been used as an excipient in the preparation of microemulsions (21) and emulsion filled gels (22), while fenofibrate has been formulated as a model drug with low aqueous solubility (0.3 mg/L) (23) and high lipophilicity (logP 5.1) (24) in SEDDS (25,26,27), in solid self-emulsifying drug delivery systems (S-SEDDS) (28,29,30,31), in self-microemulsifying drug delivery systems (SMEDDS) (32,33), and in SNEDDS (34,35,36,37,38).
As a continuation of our earlier work, in which SNEDDS formulations of fenofibrate containing different long-chain triglycerides (rice bran oil or corn oil) and different surfactants (Kolliphor RH40 or Kolliphor EL) were developed, optimized, and thoroughly characterized in terms of their physicochemical properties and biocompatibility (39), the purpose of the current study was to investigate the oral performance of the respective SNEDDS formulations. In order to efficiently simulate in vitro the physiological oral drug absorption process, a simple two-step combined digestion-permeability assessment method was adopted. Specifically, the solubilization capacity of the SNEDDS formulations was initially assessed in vitro using the static digestion model, followed by a consecutive ex vivo intestinal permeability study of the predigested SNEDDS using the non-everted gut sac model. The data of the combined in vitro/ex vivo studies were then compared with the in vivo bioavailability data obtained after the oral administration of the different SNEDDS formulations in rats.
MATERIALS AND METHODS
Materials
Corn oil (CO), l-α-phosphatidylcholine [from egg yolk, Type XVI-E, ≥ 99% by TLC, lyophilized powder], maleic acid (purity 99%), sodium taurocholate hydrate, pancreatin [8 × United States Pharmacopeia (USP) specifications], 4-bromophenylboronic acid (4-BBBA, ≥ 95.0%), fenofibrate, fenofibric acid (FA), sulfasalazine (used as internal standard), formic acid, and dichloromethane were purchased from Sigma Aldrich (St Louis, MO). Diethylene glycol monoethyl ether (Transcutol HP) and glyceryl monolinoleate (Maisine CC) were kindly gifted by Gattefossé (St. Priest, France). Polyoxyl 40 hydrogenated castor oil (Kolliphor RH40) and polyoxyl castor oil (Kolliphor EL) were kindly supplied by BASF (Ludwigshafen, Germany). Rice bran oil (RBO) was extracted based on a previously described method (39). Acetonitrile, methanol, and water (HPLC grade) were supplied by VWR chemicals (Vienna, Austria).
Preparation of the SNEDDS Formulations
The SNEDDS formulation, comprising 30% w/w rice bran oil or corn oil, 30% w/w Maisine CC, 30% w/w Kolliphor RH40 or Kolliphor EL, and 10% w/w Transcutol HP, was prepared based on previously developed and characterized compositions (39) (Table I). Briefly, Maisine CC was mixed with RBO or CO, followed by the addition of Kolliphor RH40 (heated at 50°C) or Kolliphor EL and Transcutol HP. Fenofibrate-loaded SNEDDS were prepared at a final drug concentration of 30 mg/g of each formulation. All formulations represent class IIIA lipid-based drug delivery systems (LbDDS III) according to the lipid formulation classification system (LFCS) (40).
Thermodynamic Stability and Dispersibility Studies
Thermodynamic stability studies were conducted to assess the effect of temperature variation on SNEDDS stability (41). In specific, the SNEDDS were initially subjected to six consecutive heating-cooling cycles between 45 and 4°C (48 h storage at each temperature). Formulations that were stable at the end of the heating-cooling step were centrifuged at 3500 rpm for 30 min. The SNEDDS that did not show any phase separation were further subjected to three consecutive freeze-thaw cycles between − 20°C and + 25°C (48h storage at each temperature). Formulations passing the thermodynamic stability tests were subjected to the dispersibility test in order to evaluate their self-emulsification efficacy. In particular, 1 mL of each formulation was dispersed in 500 mL of water at 37 ± 0.1°C using a USP XXII dissolution apparatus 2 with a paddle rotation speed of 50 rpm. Visual assessment of the in vitro performance of the SNEDDS was based on the following grading system:
-
Grade A: Rapidly forming (within 1 min) nanoemulsion, having a clear or bluish appearance.
-
Grade B: Rapidly forming, slightly less clear emulsion, having a bluish-white appearance.
-
Grade C: Fine milky emulsion that formed within 2 min.
-
Grade D: Dull, grayish-white emulsion having a slightly oily appearance that is slow to emulsify (longer than 2 min).
-
Grade E: Formulation, exhibiting either poor or minimal emulsification with large oil globules present on the surface.
Particle Size Measurements
Particle size measurement of the SNEDDS formulations was performed on a Malvern Zetasizer (Nano-ZS analyzer, Malvern, UK) at 25°C. The SNEDDS formulations (0.1 g) were dispersed in Milli-Q water (1 mL) and vortexed to ensure thorough homogenization prior to measurements.
Transmission Electron Microscopy
Morphological evaluation of the SNEDDS formulations was performed using Transmission electron microscopy. The SNEDDS were dispersed in Milli-Q water at a 1:100 dilution ratio and a 10 μL droplet of each sample was deposited on carbon film-coated grids and dried at ambient temperature. Samples were observed with TEM Jeol 2100 operated at 200 kV.
In Vitro Dissolution Studies in Simulated Gastric Fluid
In vitro dissolution studies were performed in 900 mL simulated gastric fluid (SGF) (USP apparatus II) at 37°C and at 50 rpm. Six hundred milligrams of each SNEDDS formulation (corresponding to 18 mg of fenofibrate) were introduced in hard gelatin capsules (size 0), while an equivalent quantity of pure fenofibrate was used as the control. Samples (1 mL) were withdrawn and replaced with an equal amount of preheated medium. Fenofibrate quantification in the samples was performed with HPLC analysis (“Fenofibrate and Fenofibric Acid Quantification” section).
Preparation of Lipolysis Medium
In vitro static lipolysis experiments were conducted in fasted state simulated intestinal fluid version 2 (FaSSIF-V2) medium (pH 6.5), based on a previously reported method by Kilic and Dressman (2013) in the presence of pancreatin extract (8 x USP specification) (7). For the preparation of FaSSIF-V2 medium, sodium chloride (4.01 g), maleic acid (3.32 g), and sodium hydroxide (2.1 g) were dissolved in 900 mL demineralized water at pH 6.5. Sodium taurocholate (1.67 g) was then dissolved in 100 mL of the medium in a round bottom flask followed by the addition of 0.5 mL lecithin solution in dichloromethane (3.15 g/10 mL), resulting in the formation of an emulsion. The organic solvent was removed by rotary evaporation at 40°C under vacuum and a 100-mbar vacuum was applied during the final step to induce the formation of a micellar solution. After cooling down at ambient temperature, the micellar solution was returned back to the initial bile salt solution in which calcium chloride (0.56 g) was dissolved, followed by the addition of 20 mL of pancreatin extract. To prepare pancreatin extract, porcine pancreatin (6.25 g) was mixed with 20 mL demineralized water and centrifuged at 2000×g and 37°C for 7 min. The supernatant was immediately introduced in the medium followed by pH adjustment to 6.5. The final volume was also adjusted to 1 L by adding demineralized water.
In Vitro Lipolysis Experiments
The in vitro lipolysis experimental setup using the static digestion model was based on Kilic and Dressman (2013) with minor modifications (7). The lipolysis process was initiated upon the addition of 10 mL of the medium in vials containing 0.1 g of the SNEDDS formulations. The vials were briefly vortexed to enable homogenous dispersion of the formulation in the medium and were incubated at 37°C on an orbital shaker. Samples (200 μL) were periodically withdrawn and immediately inhibited with the addition of 5 μL/mL lipolysis inhibitor (1 M 4-BBBA in methanol). All samples were then centrifuged at 17,000×g for 30 min. Fenofibrate quantification in the dispersed colloidal aqueous phase was performed after dilution with acetonitrile with HPLC analysis (“Fenofibrate and fenofibric acid quantification”).
Ex Vivo Permeability Study of the Predigested Fenofibrate SNEDDS Using the Non-everted Gut Sac Method
The non-everted gut sac method was used for the ex vivo permeability assessment of the predigested fenofibrate SNEDDS formulations (42). Wistar rats were fasted overnight with access to water ad libitum. The next day animals were sacrificed by cervical dislocation and the small intestine was dissected and purged with ice-cold buffered saline from the one end to thoroughly cleanse it. In order to adequately simulate the absorption process, the intestine was filled with the fenofibrate containing SNEDDS formulations (equivalent to 3 mg of fenofibrate) immediately after the completion of the in vitro digestion process in FaSSIF-V2 medium. The tissue was then successively tied with silk suture to form multiple sacs (ca. 6 cm in length), obviating intestinal regions containing Peyer’s patches. Intestinal sacs were placed in 10 mL of Ringer’s solution in glass containers. The studies were conducted in quadruplicates in an orbital shaking water bath under mild agitation and samples (0.5 mL) were withdrawn at 30, 60, 90, 120, and 150 min and replaced with an equal volume of medium. Samples were centrifuged at 10,000×g (Eppendorf centrifuge, 5417C) for 10 min and the supernatants were analyzed by HPLC (“Fenofibrate and Fenofibric Acid Quantification” section). Apparent permeability was calculated as the amount (μg) of fenofibric acid (FA), (the active metabolite of fenofibrate hydrolyzed by tissue esterases), permeation per intestinal mucosa surface area (cm2), according to Eq. (1):
where ΔQ/Δt is the flux across the intestinal sac (μg/min), A is the surface area of the sac (cm2) and C0 is the initial drug concentration (μg/mL). The mucosal surface area of the rectangular shape intestinal sacs was calculated by measuring the dimensions (i.e. width and length) after they were cut open.
In Vivo Pharmacokinetic Study
Male Wistar rats (3 months old) weighing 207 ± 11 g were used in the present study. The animals were housed in individual cages at controlled temperature (20–22°C) and humidity (50–70% RH) conditions in the authorized animal facilities of the Laboratory of Animal Physiology at the School of Veterinary Medicine of the Aristotle University of Thessaloniki. Animals were fasted overnight with free access to water. On the day of the experiment, the SNEDDS formulations were dispersed in Milli-Q water (at a 1:10 ratio) and immediately administered to the animals (n = 4) using an oral gavage (Instech Laboratories Inc., Plymouth Meeting, PA, USA). All animals received a dose of 10 mg/kg fenofibrate. Eight hours after oral dosing animals were allowed to standard feed. Blood samples (200 μL) were collected from the tail vein with a 23G needle, transferred to sodium citrate tubes and centrifuged at 2000×g for 10 min (4°C). Plasma was collected and stored at − 80°C till HPLC analysis (“Fenofibrate and Fenofibric Acid Quantification” section). The experimental protocol was approved by the Institutional Bioethics Committee for Animal Experimentation. The study was conducted in accordance with the EU Commission Directive 2010/63/EU for animal experimentation and complied with the Protocol on the Protection and Welfare of Animals, the regulations of the National Bioethics Committee and the Article 3 of the Presidential Decree 160/1991, regarding the protection of experimental animals.
Fenofibrate and Fenofibric Acid Quantification
Bioanalytical Procedure (Sample Preparation, Instrumentation and Analytical Conditions)
All samples were elaborated using a simple one-step protein precipitation. Briefly, an aliquot of 10 μL of ISTD solution (200 μg mL−1 sulfasalazine in methanol) was added to 50 μL plasma sample and vortexed for 30 s. Then, an aliquot of 140 μL ice-cold acetonitrile was added to facilitate efficient protein precipitation. After vortex-mix for 30 s the samples were centrifuged at 4000×g for 10 min and subsequently a volume of 100 μL of the supernatant were transferred to HPLC vial insert and analyzed.
Chromatographic analyses were conducted on a Shimadzu HPLC system consisted of two LC-20AD low pressure gradient pumps, an SIL-20AC HT thermostated autosampler, a CTO-20AC thermostated column compartment and a SPD-20A PDA detector (Kyoto, Japan). The control of the instrument and the data handling were carried out via the LabSolutions software (version 5.42 SP3). All separations were performed on an analytical column Hibar Lichrosorb C18 (125 × 4.6 mm id, 5 μm) (Merck). The mobile phases A and B were consisted of 0.1% v/v formic acid aqueous solution and 0.1% v/v formic acid in methanol, respectively. The gradient elution steps included an initial content of 50% B for 3 min, linear increase up to 90% in 20 min and then change to the initial composition (50% B) in 5 min followed by a column equilibrium period of 10 min to obtain reproducible separations. The flow rate was kept constant at 1 mL min−1 throughout the analysis while the injection volume was 20 μL. The column was thermostated at 30°C and the analytes were monitored spectrophotometrically at 290 nm. The processed samples were kept at 4°C in the autosampler tray.
The developed analytical method was validated in terms of selectivity, linearity, accuracy and precision according to the USFDA guidelines for the bioanalytical method. The linearity of the HPLC method was assessed between 0.1–1 and 1–40 μg mL−1 for both fenofibrate and FA with r > 0.9959 in all cases. The lower limits of quantification (LLOQ) were found to be 100 ng mL−1 for both analytes and determined by the lowest concentration measured with a precision and accuracy of 20% or less. The accuracy (expressed as % recovery) was ranged between 99.6–116.9%. The obtained validation data were within the acceptable range and thus the method was judged to be suitable for its purpose.
Pharmacokinetic Data Analysis and Statistics
The pharmacokinetic parameters (AUC: area under the plasma concentration-time curve from t = 0 h to t = 24 h, Cmax: maximum plasma concentration of the drug, Tmax: time to reach Cmax) were determined by non-compartmental analysis using the PKsolver program (43). The data are presented as means ± standard deviation. Analysis of variance (ANOVA) and Tukey’s multiple comparisons test were used to evaluate statistically significant differences between groups. The significance level was set at 0.05.
RESULTS
Thermodynamic Stability and Dispersibility Studies
The optimized SNEDDS formulations that have been developed in our previous study (39) were subjected to thermodynamic stability and dispersibility studies to further confirm their stability under thermal stress. All SNEDDS formulations passed both the thermodynamic and the dispersibility tests (Table II) and were qualified to further studies.
Particle Size Measurements and Morphological Assessment
The emulsion particle size of the different SNEDDS formulations was measured upon SNEDDS dispersion in Milli-Q water. All formulations yielded monomodal particle size distributions with narrow polydispersity indices (Fig. 1). The smaller mean particle size was observed for RBO-EL [41.8 ± 0.6 nm (PDI: 0.116)] followed by CO-EL [43.2 ± 0.3 nm (PDI: 0.133)], RBO-RH40 [54.2 ± 0.5 nm (PDI: 0.143)] and CO-RH40 [60.9 ± 0.5 nm (PDI:0.194)]. Morphological assessment of the diluted SNEDDS with TEM revealed spherical oil droplets with droplet sizes being in close proximity to the ones obtained during DLS analysis (Fig. 2).

Emulsion particle size distributions of the SNEDDS formulations after dispersion in Milli-Q water at a 1:10 dilution ratio (n = 3, ± S.D.)

Transmission electron microscopy (TEM) images of the a RBO-RH40, b CO-RH40, c RBO-EL, and d CO-EL SNEDDS formulations after dispersion in Milli-Q water at a 1:100 dilution ratio. CO: corn oil, RBO: rice bran oil, RH40: Kolliphor RH40, EL: Kolliphor EL
Fenofibrate Quantification During In Vitro Dissolution in SGF and Lipolysis in FaSSIF
The in vitro dissolution profiles of fenofibrate from the different SNEEDS formulations are shown in Fig. 3. Complete fenofibrate dissolution was observed for all SNEDDS within 15 min, compared with pure fenofibrate for which drug levels in the dissolution medium were undetectable throughout the timespan of the experimental procedure. The amount of fenofibrate quantified in the dispersed aqueous micellar phase of the lipolysis samples for all SNEDDS formulations is presented in Fig. 4a–d. The data demonstrated that the drug was found in the dispersed aqueous micellar phase, enabling solubilization of high amounts of fenofibrate (> 85%) throughout the lipolysis process. No statistically significant differences were observed between all LbDDS III, however, a trend towards higher drug solubilization was observed for the CO-RH40 SNEDDS formulation during in vitro lipolysis.

In vitro dissolution profiles of pure fenofibrate and fenofibrate SNEDDS formulations in SGF at 37°C (n = 3, ± S.D.)

Fenofibrate distribution (%) in the dispersed aqueous micellar phase during the lipolysis of the a RBO-RH40, b CO-RH40, c RBO-EL, and d CO-EL SNEDDS formulations (n = 3, ± S.D.). CO: corn oil, RBO: rice bran oil, RH40: Kolliphor RH40, EL: Kolliphor EL
Ex Vivo Intestinal Permeability Studies
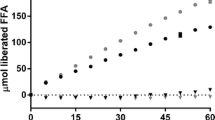
The cumulative permeation of FA across intestinal tissue was determined for the different fenofibrate-loaded predigested SNEDDS using the non-everted gut sac method (Fig. 5a). The corresponding apparent permeability values are presented in Fig. 5b. A time-dependent increase in FA permeation was observed for all SNEDDS formulations across rat intestine. Similar permeability profiles were obtained for both CO SNEDDS formulations, as well as for the RBO-RH40 formulation. On the contrary, a statistically significant enhancement in FA permeation was observed for the RBO-EL SNEDDS formulation (P < 0.05), which demonstrated at least a 2-fold increase in Papp [20.80 ± 2.54 (×10−5 cm/min)], compared with the respective values of the other tested SNEDDS formulations [CO-RH40: 9.00 ± 4.48 (×10−5 cm/min), CO-EL: 8.83 ± 1.17 (×10−5 cm/min), RBO-RH40: 7.50 ± 1.17 (×10−5 cm/min)] designating its superiority in enhancing drug transport ex vivo. The steady state flux values were calculated to be 0.062 ± 0.007 μg/min/cm2 for the RBO-EL, 0.027 ± 0.013 μg/min/cm2 for the CO-RH40, 0.026 ± 0.003 μg/min/cm2 for the CO-EL and 0.022 ± 0.004 μg/min/cm2 for the RBO-RH40 SNEDDS formulation.

Ex vivo permeability profiles of FA across rat intestine from the digested SNEDDS formulations using the non-everted gut sac method (n = 4, ± S.D.). CO: corn oil, RBO: rice bran oil, RH40: Kolliphor RH40, EL: Kolliphor EL
In Vivo Pharmacokinetic Study
The mean plasma concentration-time profiles of FA following oral administration of the SNEDDS formulations comprising different oils (rice bran oil and corn oil) and surfactants (Kolliphor EL and Kolliphor RH40) at a drug dose of 10 mg/kg were compared and are shown in Fig. 6. The corresponding pharmacokinetic parameters were calculated by non-compartmental analysis and are summarized in Table III.

Plasma concentration-time profile of FA following oral administration of the fenofibrate-loaded SNEDDS formulations (10 mg/kg) in male Wistar rats (n = 4, ± S.D.). CO: corn oil, RBO: rice bran oil, RH40: Kolliphor RH40, EL: Kolliphor EL
The AUC0 → 24 and Cmax values obtained after the oral administration of the fenofibrate-loaded SNEDDS formulations followed the order RBO-EL > CO-RH40 > RBO-RH40 ≥ CO-EL. The extent of oral bioavailability was the highest in the case of RBO-EL formulation and in close agreement with the results obtained during the ex vivo intestinal permeability of FA from the predigested SNEDDS, showing a 1.26 to 1.72-fold enhancement, compared with the rest of the tested SNEDDS formulations. Similar pharmacokinetic profiles were obtained for CO-EL, CO-RH40, and RBO-RH40 SNEDDS, whereas a statistically significant difference was only observed for the Cmax of the CO-RH40 SNEDDS (p < 0.05), compared with the other formulations. Maximum plasma concentrations were attained within approximately 2 h for both RBO-EL and CO-RH40 SNEDDS, whereas slightly longer Tmax values (3 h) were observed for CO-EL and RBO-RH40 SNEDDS.
DISCUSSION
The aim of the current study was to investigate the effect of different SNEDDS formulations, that we have previously developed, optimized, and thoroughly characterized (39), on their ability to enhance the oral absorption of a BCS class II drug. In pursuance of a simplified approach to more adequately simulate the physiological fate of LbDDS upon oral ingestion, a consecutive in vitro digestion (using the static model) and ex vivo intestinal permeability (using the non-everted gut sac model) assessment of the predigested SNEDDS was adopted, prior to the evaluation of their in vivo performance.
In the current study, the SNEDDS formulations were prepared using two different oils (rice bran oil or corn oil) and two different surfactants (Kolliphor RH40 or Kolliphor EL) and fenofibrate were chosen as a model lipophilic active compound. In vitro lipolysis was performed in FaSSIF-V2 medium using the static digestion model that has been established as an alternative to pH-stat lipolysis, providing higher throughput for dispersion and lipolysis assessment of LbDDS (7). All tested SNEDDS formulations demonstrated adequate thermodynamic stability. Dispersion of the SNEDDS in Milli-Q water resulted in emulsions with monomodal particle size distributions in the range below 100 nm, as also visualized with TEM, with RBO-EL yielding the smallest mean particle size. Fenofibrate showed complete dissolution from all SNEDDS in SGF within 15 min, while during SNEDDS digestion, fenofibrate demonstrated a major distribution in the aqueous phase throughout the lipolysis process. Even though no statistically significant differences were observed in the solubilization capacity between the different SNEDDS compositions, CO-RH40 showed a tendency towards retaining higher amounts of the drug in solubilized form. Based on that observation one would expect a similar trend to be captured in the in vivo experiments, as well.
However, no clear correlation could be obtained between the in vitro solubilization capacity of the SNEDDS and their efficacy in enhancing the oral absorption of fenofibrate in vivo, since the highest FA bioavailability was achieved after the oral administration of the RBO-EL SNEDDS formulation. The inability of in vitro digestion models to consistently predict the oral performance of SNEDDS containing fenofibrate has been recently highlighted, underlying that enhanced drug solubilization is not always an indication of improved oral absorption (38), supporting the findings of the present work. In previous studies, the in vitro/in vivo correlation between SEDDS digestibility and extent of oral drug absorption was tested to elucidate the effect of Kolliphor™ surfactants on fenofibrate solubilization and bioavailability (44). RH40-containing SEDDS were found to be less digestible during in vitro lipolysis studies compared with the respective EL-containing SEDDS formulations, thus retaining a high drug solubilization capacity, nevertheless, a higher oral bioavailability was observed for the EL-containing SEDDS, similarly to the results obtained in the current study for the RBO-EL SNEDD. At the same time, the RBO-EL SNEDD formulation yielded the smallest particle size after dispersion in water, which generally has been correlated with increased drug absorption compared with SNEDDS with larger particle sizes (45). In addition to that, it has been previously shown that the fatty acid composition of vegetable oils, employed as the lipid carrier of lipophilic compounds, is a determinant factor of drug micellization prior to intestinal absorption, with the rate of drug absorption depending on micellar fatty acid hydrophobicity and extent of saturation (45). In particular, Nidhi B. et al. (2011) showed that among different vegetable oils evaluated as lutein lipid carriers, those having the highest oleic/linoleic (18:1/18:2) fatty acid content enhanced the percentage of lutein micellization after in vitro digestion, which was further ranked in the order olive oil > groundnut oil > rice bran oil > sunflower oil > corn oil > soybean oil > palm oil in close agreement to the ranking order obtained in the current study for the RBO-EL formulation (46). This finding further signifies the potential of RBO as an excipient for enhancing the oral absorption of BCS class II compounds and the significance of biowaste valorization for pharmaceutical applications, as well.
CONCLUSION
The findings of the current study demonstrate the potential of sustainable use of agro-industrial waste in pharmaceutical formulations with the aim to enhance the oral absorption of a BCS II active compound. The ex vivo permeability assessment of different predigested SNEDDS formulations, comprising different lipidic vehicles (LCT) and different surfactants showed that combination of RBO with Kolliphor EL increased the oral bioavailability of fenofibrate in rats, in agreement with the findings of the combined digestion-permeability study utilizing the in vitro static digestion model and the non-everted gut sac permeability model.
References
Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov Engl. 2007;6:231–48.
Rehman FU, Shah KU, Shah SU, Khan IU, Khan GM, Khan A. From nanoemulsions to self-nanoemulsions, with recent advances in self-nanoemulsifying drug delivery systems (SNEDDS). Expert Opin Drug Deliv Engl. 2017;14:1325–40.
Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12:1561–72.
Thomas N, Holm R, Rades T, Müllertz A. Characterising lipid lipolysis and its implication in lipid-based formulation development. AAPS J. 2012;14:860–71.
Norinder U, Haeberlein M. Calculated molecular properties and multivariate statistical analysis in absorption prediction. Drug Bioavailabil. 2003:358–405.
Mosgaard MD, Sassene P, Mu H, Rades T, Müllertz A. Development of a high-throughput in vitro intestinal lipolysis model for rapid screening of lipid-based drug delivery systems. Eur J Pharm Biopharm. 2015;94:493–500.
Kilic M, Dressman J. A simplified method to screen for in-vivo performance of oral lipid formulations. J Pharm Pharmacol. 2014;66:615–23.
Berthelsen R, Klitgaard M. In vitro digestion models to evaluate lipid based drug delivery systems; present status and current trends. Adv Drug Deliv Rev. 2019;142:35–49.
Keemink J, Mårtensson E, Bergström CAS. Lipolysis-permeation setup for simultaneous study of digestion and absorption in vitro. Mol Pharm. 2019;16:921–30.
Dahan A, Hoffman A. The effect of different lipid-based formulations on the oral absorption of lipophilic drugs: the ability of in vitro lipolysis and consecutive ex vivo intestinal permeability data to predict in vivo bioavailability in rats. Eur J Pharm Biopharm. 2007;67:96–105.
Bibi HA, Holm R, Bauer-Brandl A. Simultaneous lipolysis/permeation in vitro model, for the estimation of bioavailability of lipid-based drug delivery systems. Eur J Pharm Biopharm. 2017;117:300–7.
Pandey V, Kohli S. Lipids and surfactants: the inside story of lipid-based drug delivery systems. Crit Rev Ther Drug Carrier Syst. 2018;35:99–155.
Sohail M, Rakha A, Butt MS, Iqbal MJ, Rashid S. Rice bran nutraceutics: a comprehensive review. Crit Rev Food Sci Nutr. 2017;57:3771–80.
Ricciardi P, Cillari G, Carnevale Miino M, Collivignarelli MC. Valorization of agro-industry residues in the building and environmental sector: a review. Waste Manag Res J Int Solid Wastes. 2020:734242X20904426.
Pal YP, Pratap AP. Rice bran oil: a versatile source for edible and industrial applications. J Oleo Sci. 2017;66:551–6.
Ortheofer F. Rice bran oil, Bailey’s industrial oil and fat products. 6th ed. New Jersey: John Wiley & sons; 2005. p. 465–89.
Sirithunyalug B, Saenjum C, Charumanee S, Sivamaruthi BS, Chaiyasut C, Sirithunyalug J, et al. Development of colorectal-targeted dietary supplement tablets containing natural purple Rice bran oil as a colorectal chemopreventive. Nutrients. 2018;10.
Devarajan S, Singh R, Chatterjee B, Zhang B, Ali A. A blend of sesame oil and rice bran oil lowers blood pressure and improves the lipid profile in mild-to-moderate hypertensive patients. J Clin Lipidol. 2016;10:339–49.
Hunthayung K, Klinkesorn U, Hongsprabhas P, Chanput W. Controlled release and macrophage polarizing activity of cold-pressed rice bran oil in a niosome system. Food Funct. 2019;10:3272–81.
Ghatak SB, Panchal SS. Anti-diabetic activity of oryzanol and its relationship with the anti-oxidant property. Int J Diabetes Dev Ctries. 2012;32:185–92.
Wuttikul K, Boonme P. Formation of microemulsions for using as cosmeceutical delivery systems: effects of various components and characteristics of some formulations. Drug Deliv Transl Res. 2016;6:254–62.
Nourbehesht N, Shekarchizadeh H, Soltanizadeh N. Investigation of stability, consistency, and oil oxidation of emulsion filled gel prepared by inulin and rice bran oil using ultrasonic radiation. Ultrason Sonochem. 2018;42:585–93.
Granero GE, Ramachandran C, Amidon GL. Dissolution and solubility behavior of fenofibrate in sodium lauryl sulfate solutions. Drug Dev Ind Pharm. 2005;31:917–22.
Persson LC, Porter CJH, Charman WN, Bergström CAS. Computational prediction of drug solubility in lipid-based formulation excipients. Pharm Res. 2013;30:3225–37.
Pestieau A, Lebrun S, Cahay B, Brouwers A, Streel B, Cardot J-M, et al. Evaluation of different in vitro dissolution tests based on level A in vitro–in vivo correlations for fenofibrate self-emulsifying lipid-based formulations. Eur J Pharm Biopharm. 2017;112:18–29.
Tran T, Siqueira SDVS, Amenitsch H, Müllertz A, Rades T. In vitro and in vivo performance of monoacyl phospholipid-based self-emulsifying drug delivery systems. J Control Release. 2017;255:45–53.
Pestieau A, Krier F, Brouwers A, Streel B, Evrard B. Selection of a discriminant and biorelevant in vitro dissolution test for the development of fenofibrate self-emulsifying lipid-based formulations. Eur J Pharm Sci. 2016;92:212–9.
Shazly G, Mohsin K. Dissolution improvement of solid self-emulsifying drug delivery systems of fenofibrate using an inorganic high surface adsorption material. Acta Pharma. 2015;65:29–42.
Quan G, Wu Q, Zhang X, Zhan Z, Zhou C, Chen B, et al. Enhancing in vitro dissolution and in vivo bioavailability of fenofibrate by solid self-emulsifying matrix combined with SBA-15 mesoporous silica. Colloids Surf B Biointerfaces. 2016;141:476–82.
Kanaujia P, Ng WK, Tan RBH. Solid self-emulsifying drug delivery system (S-SEDDS) for improved dissolution rate of fenofibrate. J Microencapsul. 2014;31:293–8.
Shah AV, Serajuddin ATM. Development of solid self-emulsifying drug delivery system (SEDDS) I: use of poloxamer 188 as both solidifying and emulsifying agent for lipids. Pharm Res. 2012;29:2817–32.
Lee DW, Marasini N, Poudel BK, Kim JH, Cho HJ, Moon BK, et al. Application of box-Behnken design in the preparation and optimization of fenofibrate-loaded self-microemulsifying drug delivery system (SMEDDS). J Microencapsul. 2014;31:31–40.
Sunazuka Y, Ueda K, Higashi K, Tanaka Y, Moribe K. Combined effects of the drug distribution and mucus diffusion properties of self-microemulsifying drug delivery systems on the oral absorption of fenofibrate. Int J Pharm. 2018;546:263–71.
Ren S, Mu H, Alchaer F, Chtatou A, Müllertz A. Optimization of self nanoemulsifying drug delivery system for poorly water-soluble drug using response surface methodology. Drug Dev Ind Pharm. 2013;39:799–806.
Mohsin K, Alamri R, Ahmad A, Raish M, Alanazi FK, Hussain MD. Development of self-nanoemulsifying drug delivery systems for the enhancement of solubility and oral bioavailability of fenofibrate, a poorly water-soluble drug. Int J Nanomedicine. 2016;11:2829–38.
Alshamsan A, Kazi M, Badran MM, Alanazi FK. Role of alternative lipid excipients in the design of self-nanoemulsifying formulations for fenofibrate: characterization, in vitro dispersion, digestion and ex vivo gut permeation studies. Front Pharmacol. 2018;9:1219.
Tran T, Bønløkke P, Rodríguez-Rodríguez C, Nosrati Z, Esquinas PL, Borkar N, et al. Using in vitro lipolysis and SPECT/CT in vivo imaging to understand oral absorption of fenofibrate from lipid-based drug delivery systems. J Control Release. 2020;317:375–84.
Thomas N, Richter K, Pedersen TB, Holm R, Müllertz A, Rades T. In vitro lipolysis data does not adequately predict the in vivo performance of lipid-based drug delivery systems containing fenofibrate. AAPS J. 2014;16:539–49.
Eleftheriadis GK, Mantelou P, Karavasili C, Chatzopoulou P, Katsantonis D, Irakli M, et al. Development and characterization of a self-nanoemulsifying drug delivery system comprised of rice bran oil for poorly soluble drugs. AAPS PharmSciTech. 2019;20:78.
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29:278–87.
Shafiq S, Shakeel F, Talegaonkar S, Ahmad FJ, Khar RK, Ali M. Development and bioavailability assessment of ramipril nanoemulsion formulation. Eur J Pharm Biopharm. 2007;66:227–43.
Kontogiannidou E, Karavasili C, Kouskoura MG, Filippousi M, Van Tendeloo G, Andreadis II, et al. In vitro and ex vivo assessment of microporous faujasite zeolite (NaX-FAU) as a carrier for the oral delivery of danazol. J Drug Deliv Sci Technol. 2019;51:177–84.
Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Prog Biomed. 2010;99:306–14.
Berthelsen R, Holm R, Jacobsen J, Kristensen J, Abrahamsson B, Müllertz A. Kolliphor surfactants affect solubilization and bioavailability of fenofibrate. Studies of in vitro digestion and absorption in rats. Mol Pharm. 2015;12:1062–71.
Hussain A, Singh SK, Singh N, Prasad Verma PR. In vitro–in vivo–in silico simulation studies of anti-tubercular drugs doped with a self-nanoemulsifying drug delivery system. RSC Adv. 2016;6:93147–61.
Nidhi B, Baskaran V. Influence of vegetable oils on micellization of lutein in a simulated digestion model. J Am Oil Chem Soc. 2011;88:367–72.
Acknowledgments
Gattefosse (St. Priest, France) and BASF (Ludwigshafen, Germany) are greatly acknowledged for the donation of the formulation excipients. The authors thank Dr. Maria Kollia of the Laboratory of Electron Microscopy and Microanalysis, School of Natural Sciences, University of Patras, Greece, and Prof. Nikolaos Bouropoulos from the Department of Materials Science, University of Patras, Greece, for the TEM images.
Funding
The current work has been supported by the “Sustainable techno-economic solutions for the agricultural value chain” Waste-7-2015 topic H2020 690142 project (AGROCYCLE).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Karavasili, C., Andreadis, I.I., Tsantarliotou, M.P. et al. Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) Containing Rice Bran Oil for Enhanced Fenofibrate Oral Delivery: In Vitro Digestion, Ex Vivo Permeability, and In Vivo Bioavailability Studies. AAPS PharmSciTech 21, 208 (2020). https://doi.org/10.1208/s12249-020-01765-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-020-01765-2