
Abstract
The USP Apparatus 3 is a compendial dissolution Apparatus that has been mainly used to assess the performance of modified-release drug products. However, this Apparatus can be applied to dissolution testing of immediate-release tablets as well, with several advantages such as lower consumption of dissolution media, reduced setup time in quality control routine, and minimized hydrodynamic issues. In this work, three immediate-release (IR) tablets containing antihypertensive drugs of different Biopharmaceutic Classification System (BCS) classes were evaluated in order to assess the possible interchangeability between the official dissolution method using typical USP Apparatus 1 or 2 and the proposed methods using USP Apparatus 3. Depending on the selection of the appropriate operational conditions, such as dip rate and sieve mesh size, it was observed that USP Apparatus 3 could provide similar dissolution profiles compared to USP Apparatus 1 or 2 to the drug products tested. In addition, USP Apparatus 3 avoided conning issues related to USP Apparatus 2. The successful application of USP Apparatus 3 in dissolution tests for IR drug products depends on the definition of specific test conditions for each product, considering all the equipment variables, as well as drug and formulation characteristics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Dissolution testing is widely used as a quality control tool to assess the performance of solid oral dosage forms. A meaningful dissolution test should be representative of the physiological conditions that the dosage form will be exposed in order to predict its in vivo behavior (1,2,3). Currently, the United States Pharmacopeia (USP) Apparatus 1 and 2 (basket and paddle, respectively) are the most used dissolution Apparatus in official monographs of the main pharmacopoeias from all over the world.
A review of Food and Drug Administration (FDA) dissolution methods database was recently published and, according to it, paddle is the most common Apparatus adopted in pharmacopoeial monographs. This Apparatus is recommended for approximately 70% of the dissolution methods, and it is considered the first choice for dissolution testing of immediate-release (IR) solid dosage forms. In addition, approximately 17% of dissolution methods recommend the use of basket, and 13% recommend other Apparatus (USP Apparatus 3 to 7) (4).
Despite being the most applied in pharmacopoeial dissolution tests, USP Apparatus 1 and 2 have several disadvantages. Dissolution testing performed with USP Apparatus 1 may suffer interference and obstruction of the basket sieve, as well as the drug particles may be expelled and float to the surface of the dissolution medium or settle at the bottom of the vessel, according to their density. On the other hand, the coning effect, characterized by the formation of a cone of powder/granules at the bottom of the vessel, is commonly observed when using USP Apparatus 2. This condition is particularly critical for drug products containing poorly soluble drugs dispersed in a high percentage of insoluble excipients or tests conditions using low paddle rotation, e.g., 50 rpm, due to the poor agitation capacity of the system. Particles in the cone are subjected to lower agitation speed, which may reduce the drug dissolution and present highly variable results (5,6).
Due to the necessity to establish an in vitro-in vivo correlation by providing pharmacokinetic and mechanical conditions closer to the gastrointestinal tract in dissolution testing, USP Apparatus 3 was developed (7,8). This Apparatus was incorporated into USP in 1991, as an alternative to USP Apparatus 1 and 2 in assessment of dissolution characteristics of solid oral modified-release dosage forms (6). USP Apparatus 3 usually has six inner tubes, which mechanically traverse six rows of corresponding media-filled outer tubes. The traditional configuration utilizes a 300-mL vessel, which can be an advantage for products that require a small volume of dissolution medium (9).
USP Apparatus 3 offers the advantages of simulating changes in physiological conditions and the mechanical forces suffered by products in the gastrointestinal tract (7), and it is particularly useful for products containing poorly water-soluble drugs, modified-release technologies, and compounds that exhibit pH-dependent dissolution characteristics (1). The literature reported that USP Apparatus 3 seems to exhibit superior hydrodynamics when compared to USP Apparatus 1 and 2, due to the upward and downward movements. The reciprocal action generates a continuous fluid flow through the lower screen and allows the dosage form to move freely through the dissolution medium. Studies also demonstrated that the results in USP Apparatus 3 are not sensitive to factors such as the presence of sample collection probes or air bubbles in the dissolution medium (1,6,8,10).
Although it has been conceived for dissolution of modified-release dosage forms, USP Apparatus 3 has a significant potential for assessing the dissolution performance of IR dosage forms (6), what increases its versatility and presents advantages, such as economy of dissolution medium, due to the use of reduced volume when compared to the traditional USP Apparatus 1 and 2 (sink conditions must be guaranteed). In a perspective of quality control routine, this Apparatus can also reduce the setup time between dissolution tests. As the equipment has several rows, the vessels can be filled with media for more than one dissolution test of IR products. As soon as a dissolution test is finished, another test with a different batch or drug product can be performed, since the medium would be already placed and warmed in the same dissolution equipment.
USP 39 (11) currently presents eight drug product monographs using USP Apparatus 3 for dissolution testing, where two of them refer to IR dosage forms (liothyronine sodium tablets and hydroxyzine hydrochloride tablets). In the case of liothyronine sodium tablets, it is important to highlight that the USP Apparatus 3 is the only Apparatus recommended for the dissolution testing of the drug product.
In contrast, few studies were found focusing on the use of USP Apparatus 3 to assess the dissolution of IR drug products, or comparing it to the other compendial Apparatus (5,7,8,12,13,14,15). The more expressive work reported was conducted by Yu et al., where the authors studied the application of USP Apparatus 3 in dissolution testing of four different highly and poorly soluble drug IR products. However, the authors compared the USP Apparatus 3 to the USP Apparatus 2 for IR tablets set at a fixed rotation speed (50 rpm) for all drug products tested. Besides, only one dosage form of the tablets was tested, and information about sink conditions was not described (7).
The aim of this study was to evaluate the use of USP Apparatus 3 for dissolution testing of three antihypertensive IR drug products in comparison to the current compendial methods using both USP Apparatus 1 and 2. The drug products selected present different solubility properties according to the Biopharmaceutic Classification System (BCS) (16). Dissolution profiles of hydrochlorothiazide (a poorly soluble BCS class IV drug), captopril, and atenolol (highly soluble drugs belonging to BCS class III) were determined from commercially available tablets. In order to present a more comprehensive work, two different dosages of each drug product (lower and higher dosage) were tested and evaluated under sink conditions using both compendial and the proposed USP Apparatus 3 methods. Four different agitation speeds were tested to each drug product in USP Apparatus 3. The best operational conditions such as dips per minute (dpm) and sieve mesh size were defined for these specific drug products, in order to reach similar dissolution profiles between the USP Apparatus 1 or 2 and USP Apparatus 3 methods.
MATERIAL AND METHODS
Materials
Clorana® (hydrochlorothiazide 25 and 50 mg tablets by Sanofi – Aventis Pharmaceutical Ltd.), Atenol® (atenolol 25 and 100 mg tablets from AstraZeneca Ltd.), and Captosen® (captopril 25 and 50 mg tablets by Pharlab Pharmaceutical Industry) were purchased from local pharmacies. Hydrochloric acid, phosphoric acid, methanol, sodium acetate, ammonium acetate, and acetonitrile were all analytical or high-performance liquid chromatography (HPLC) grade.
Solubility Measurement
The equilibrium solubility of hydrochlorothiazide, atenolol, and captopril was determined by the standard shake flask method (17), using a Shaker Incubator NT-715 (Nova Técnica, Brazil). An excess of each drug was added to 10 mL of the dissolution medium described in Table I and submitted to agitation of 240 rpm for 24 h at 37.0 ± 1.0 °C. Aliquots of 1 mL were withdrawn at every 12 h and immediately filtered through a 0.45-μm polyamide membrane (Chromafil® Xtra), diluted in the respective media, and quantified by specific HPLC methods to each drug product. All experiments were performed in triplicate.
Dissolution Profiles of Captopril, Atenolol, and Hydrochlorothiazide Tablets Using Compendial Methods
Dissolution profiles of captopril (25 and 50 mg), atenolol (25 and 100 mg), and hydrochlorothiazide (25 and 50 mg) tablets were conducted by the dissolution methods specified in USP 39. Dissolution tests were performed using a Varian VK 7000 dissolution tester (USA) at constant temperature of 37 ± 0.5 °C, 900 mL of dissolution medium, and specific dissolution Apparatus and rotation speeds (Table I). Five-milliliter aliquots were withdrawn at predefined sampling time intervals without medium replacement. Samples were filtered by quantitative filter paper (Unifil® C40, 6-μm pore size) prior to analysis. The dissolution profiles were determined in triplicate. The specified test conditions for each drug product are described in Table I.
Dissolution Profiles of Captopril, Atenolol, and Hydrochlorothiazide Tablets Using USP Apparatus 3 Methods
The dissolution profiles of captopril (25 and 50 mg), atenolol (25 and 100 mg), and hydrochlorothiazide (25 and 50 mg) tablets were performed in USP Apparatus 3 (RRT10, Erweka, Germany) using 250 mL of dissolution medium at constant temperature of 37 ± 0.5 °C and different dip rates. Tablets were placed into the inner cylinders using top and bottom polypropylene sieves of specified mesh sizes. Five-milliliter aliquots were withdrawn at predefined sampling time intervals without medium replacement. Samples were filtered by quantitative filter paper (Unifil® C40, 6-μm pore size) prior to analysis. The dissolution profiles were determined in triplicate. The specified test conditions for each drug product are described in Table II.
Quantification Methods
Captopril
Quantification of captopril dissolution samples was performed by HPLC (Shimadzu Corporation, Japan) using a Phenomenex® C18 reversed phase column (5 μm, 250 × 4.6 mm) at 40 °C. The mobile phase consisted of acidified water (with phosphoric acid; pH 2.5) and methanol (40:60 v/v). The flow rate was 1.0 mL min−1 and the injection volume was 20 μL. Detection was accomplished by ultraviolet (UV) absorption at 212 nm (18). Quantification of captopril samples was performed based on the regression curve (r = 0.99).
Atenolol
Quantification of atenolol dissolution samples was performed by HPLC (Shimadzu Corporation, Japan) using a Phenomenex® C18 reversed phase column (5 μm, 250 × 4.6 mm) at 40 °C. The mobile phase consisted of 10 mM ammonium acetate buffer pH 7.0 and acetonitrile (80:20 v/v). The flow rate was 0.8 mL min−1 and the injection volume was 20 μL. Detection was accomplished by UV absorption at 275 nm (19). Quantification of atenolol was performed based on a regression curve (r = 0.99).
Hydrochlorothiazide
Quantification of hydrochlorothiazide dissolution samples was performed by UV Spectroscopy (Varian, USA) at 272 nm (USP 39, 2016), using the dissolution medium as blank solution. Quantification of hydrochlorothiazide was performed based on a regression curve (r = 0.99).
Dissolution Data Treatment
The similarity of performance between methods using the Apparatus described in USP monographs and the new proposed methods using USP Apparatus 3 was assessed by mathematical comparison of dissolution profiles.
Similarity Factor
The dissolution profiles of hydrochlorothiazide tablets were compared by the model-independent method of similarity factor (f2). It is a logarithmic transformation of the sum-squared error of differences between two formulations (test and reference) over all time points, where n is the number of time points, and Rt and Tt are the percent dissolved of the reference and test formulation, respectively, at each time point (Eq. 1).
Four data points (5, 10, 15, and 30 min) were considered to perform f2 calculations, allowing the use of only one measurement above 85% (20).
A menu-driven add-in program (DDSolver) in MS Excel computed the f2 values. According to the FDA, two dissolution profiles are similar if the f2 value is between 50 and 100 (FDA, 1997).
f2 calculations were not applicable form comparison of dissolution profiles of atenolol and captopril tablets, since these drug products demonstrated a very rapid dissolution in all conditions tested (more than 85% dissolved in 15 min) (20,21). In this case, according to the current EMA and FDA Guidelines, it is assumed that the dissolution profiles obtained by the different methods may be accepted as similar without further mathematical evaluation (21,22,23,24).
Dissolution Efficiency
In other to explore and complement the experimental data obtained under the different test conditions, an additional mathematical tool was applied. The dissolution efficiency (DE) was calculated using all data points and compared by one-way analysis of variance (ANOVA) and t test to the smaller and higher drug dosages, respectively. DE is a parameter used to characterize drug release, defined as the area under the dissolution curve (AUC) up to a certain time, expressed as a percentage of rectangle area described by 100% dissolution in the same time, where y is drug percentage dissolved at time t (Eq. 2) (25,26).
A menu-driven add-in program (DDSolver) in MS Excel computed the DE values, and the statistical analyses were performed using GraphPad Prism® v5.0 software. Differences were considered significant when p < 0.05 for a confidence level of 95%.
RESULTS AND DISCUSSION
Equilibrium Solubility of Hydrochlorothiazide, Atenolol, and Captopril
Equilibrium solubility of the drugs were determined in the media described by USP official monographs for each drug product in order to verify the attendance of sink conditions for both compendial and the proposed USP Apparatus 3 methods. Equilibrium was achieved before 24 h (less than 10% variation in drug concentration) for all drugs tested, resulting in a final drug concentration of 0.69 mg/mL of hydrochlorothiazide, 36.28 mg/mL of atenolol, and 135.90 mg/mL of captopril.
Sink conditions are defined as the use of a medium volume at least three times the volume required to form a saturated solution of drug substance (11). Equilibrium solubility measurements demonstrated that sink conditions were achieved for atenolol, captopril, and hydrochlorothiazide for both compendial and proposed USP Apparatus 3 methods. The worst case among the drugs studied was hydrochlorothiazide, a BCS class IV drug with poor aqueous solubility. However, even for the highest dosage of hydrochlorothiazide tablets, sink conditions were achieved, since the volume of 250 mL used in USP Apparatus 3 method is about 3.5 times the volume required to form a saturated solution.
Dissolution Profiles of Captopril Tablets in Different Apparatus
The dissolution profiles of captopril 25 mg tablets were conducted in USP Apparatus 1, 50 rpm, as described in USP monograph (11), and in USP Apparatus 3 at different dip rates (5, 10, 20, and 30 dpm) with a 420-μm sieve. Dissolution profiles of captopril 25 mg tablets are presented in Fig. 1.

Dissolution profiles (mean ± SD) of captopril 25 mg tablets
Table III summarizes the DE values and cumulative drug release (%) obtained to captopril 25 mg tablets.
The drug product met the requirements of pharmacopoeial dissolution specification when using the compendial method and using USP Apparatus 3 in all dip rates tested (Table III). At visual observation, the dissolution profiles obtained to USP Apparatus 1 and 3 were very similar (Fig. 1). After 5 min, more than 90% of the drug was dissolved in the medium in all conditions tested, due to the high solubility of captopril in HCl 0.01N.
An IR product has a very rapid in vitro dissolution when 85% or more of the labeled amount of the drug dissolves within 15 min (21). Therefore, the dissolution profiles obtained to the proposed USP Apparatus 3 with 5, 10, 20, or 30 dpm were considered similar to the dissolution profile obtained to the compendial method (USP Apparatus 1, 50 rpm). In addition, the DE values (Table III) of captopril dissolution profiles in USP Apparatus 3 were statistically similar to the results obtained to USP Apparatus 1, 50 rpm (p > 0.05).
In order to assess the dissolution profiles of a higher dosage drug product (50 mg tablets), the milder condition used in USP Apparatus 3 (5 dpm) for captopril 25 mg tablets was chosen. Therefore, captopril 50 mg tablets were tested in USP Apparatus 1 according to the compendial method and in USP Apparatus 3 with 5 dpm and a 420-μm sieve. Dissolution profiles of captopril 50 mg tablets are demonstrated in Fig. 2.

Dissolution profiles (mean ± SD) of captopril 50 mg tablets
As observed to captopril 25 mg, the dissolution profiles of captopril 50 mg tablets obtained using the compendial method (USP Apparatus 1, 50 rpm) and USP Apparatus 3 with 5 dpm demonstrated a very rapid in vitro dissolution, being considered similar to each other.
Captopril 50 mg tablets showed a cumulative drug release at 20 min of 99.5 ± 2.2% and 99.0 ± 2.4%, and DE values of 91.3 ± 1.3% and 91.3 ± 3.0% when using the compendial and the proposed USP Apparatus 3 methods, respectively. The DE values obtained were statistically similar (p > 0.05). The drug product also complied the pharmacopoeial dissolution specification when using the compendial and the new proposed USP Apparatus 3 methods.
Based on these findings, it was demonstrated that the proposed USP Apparatus 3 method provided similar dissolution profiles for captopril 25 mg and 50 mg tablets when compared to the compendial method.
Dissolution Profiles of Atenolol Tablets in Different Apparatus
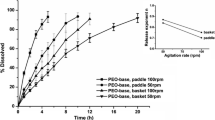
The dissolution profiles of atenolol 25 mg tablets obtained applying USP Apparatus 2, 50 rpm, and the proposed USP Apparatus 3 method at different dip rates (5, 10, 20, and 30 dpm) are presented in Fig. 3. Table IV presents the DE values and cumulative drug release (%) obtained for each condition tested, as well as the monograph specification for dissolution testing of atenolol tablets (11).

Dissolution profiles (mean ± SD) of atenolol 25 mg tablets
Atenolol 25 mg tablets met the requirements of pharmacopoeial dissolution specification to all conditions performed in USP Apparatus 2 and 3 (Table III). After only 5 min, more than 85% of atenolol was dissolved in the medium in all conditions tested, exhibiting a very rapid in vitro dissolution, as observed to captopril. Atenolol is a BCS class III drug, as captopril, and the equilibrium solubility study demonstrated the high solubility of the drug in the dissolution medium. Therefore, the dissolution profiles of atenolol 25 mg using the proposed USP Apparatus 3 method with 5, 10, 20, or 30 dpm were similar to that obtained using the compendial method (USP Apparatus 2, 50 rpm). Additionally, the DE values obtained to all conditions tested in USP Apparatus 3 were statistically similar to the result obtained when using the compendial method (p > 0.05), except for 10 dpm (p < 0.05) (Table III).
Considering the similarity of dissolution profiles achieved to atenolol 25 mg tablets, the milder condition applied in USP Apparatus 3 for this drug product was applied to the evaluation of the higher dosage (5 dpm). Therefore, atenolol 100 mg tablets were tested in USP Apparatus 2, 50 rpm, and in USP Apparatus 3 with 5 dpm and a 420-μm sieve.
Figure 4 illustrates the dissolution profiles of atenolol 100 mg tablets. Coning was observed during the dissolution test in USP Apparatus 2 at 50 rpm (images not shown). This effect is a major issue resulting from variable hydrodynamics, especially for USP Apparatus 2. It is formed due to granules or particles which have high density, resulting in a mound on the bottom of the vessel that inhibits the dissolution in the stagnant zone below the paddle (27,28).

Dissolution profiles (mean ± SD) of atenolol 100 mg tablets
Coning occurrence resulted in a more gradual release of atenolol from the 100 mg tablets when compared to atenolol 25 mg tablets at similar conditions. It was probably due to the higher dosage tablet, which has the same excipients, but an average weight fourfold higher than the smaller dosage tested, resulting in an accumulation of powder on the bottom of the vessel and a cone formation, generating considerably different results. Similar findings were reported for atenolol 100 mg tablets by Seeger et al. (2015) at 50 rpm. These authors also demonstrated that the increasing of rotation to 75 rpm minimized the coning effect and increased the drug dissolution (29).
In fact, coning may be reduced by increasing the paddle speed during the dissolution test (24). However, in the present research, this was not investigated, since the aim of the study was to compare the compendial method and the proposed USP Apparatus 3 methods, assessing the potential of USP Apparatus 3 on dissolution testing of IR drug products.
In summary, atenolol 100 mg tablets showed a cumulative drug release of 90.2 ± 7.8% at 30 min and DE value of 77.1 ± 5.4% when the compendial method was performed. Using the proposed USP Apparatus 3 method, it reached a cumulative drug release of 104.6 ± 5.3% at 30 min and a DE value of 92.6 ± 1.8%.
Even with the occurrence of coning in USP Apparatus 2, 85% of the drug was dissolved in the dissolution medium at 15 min. Using the proposed USP Apparatus 3 method, 98% of drug was dissolved at 15 min. Despite the statistically different DE values (p < 0.05), the drug product showed a very rapid in vitro dissolution behavior in both methods tested. For this reason, it is possible to conclude that the compendial method and the proposed USP Apparatus 3 method provided similar dissolution profiles for atenolol 100 mg tablets.
It is important to highlight that coning effect was not observed when using the USP Apparatus 3. This fact suggests that the proposed USP Apparatus 3 method presented a more reliable result for atenolol 100 mg tablets when compared to the compendial method, avoiding the hydrodynamic issues related to the USP Apparatus 2 method.
Dissolution Profiles of Hydrochlorothiazide Tablets in Different Apparatus
Figure 5 illustrates the dissolution profiles of hydrochlorothiazide tablets using USP Apparatus 1, 100 rpm, and USP Apparatus 3 at different dip rates. The f2, DE, cumulative drug release (%), and pharmacopoeial dissolution specification were demonstrated in Table V (10).

Dissolution profiles (mean ± SD) of hydrochlorothiazide 25 mg tablets
Considering the f2 values, none of the conditions tested in USP Apparatus 3 (5, 10, 20, and 30 dpm) with a 420-μm sieve reached dissolution profile similarity when compared to the compendial method (USP Apparatus 1; 100 rpm). The DE values for the compendial method and all conditions tested in USP Apparatus 3 were also statistically different (p < 0.05).
The selection of appropriate bottom sieve is another advantage of the USP Apparatus 3, which can vary in size and material. Typical sieves are produced in stainless steel or polypropylene and a mesh size ranging from 20 to 400. The choice of the sieve is based on its ability to retain the dosage form and allow the maximum amount of media to move through the double-ended chamber of USP Apparatus 3. During the upstroke movement of the cylinders, the drug product will contact the bottom sieve only to be suspended again on the next downstroke. This movement is particularly important to avoid the hydrodynamic issues related to the other typical compendial Apparatus, especially USP Apparatus 2 (8).
Using USP Apparatus 3 with a 420-μm sieve on the inner tube, it was observed that the remaining powder went through the sieve after tablet disintegration and settled on the bottom of the dissolution vessel, not being able to suffer the influence of the Apparatus hydrodynamics. For this reason, the dissolution of hydrochlorothiazide in USP Apparatus 3 was slower than in USP Apparatus 1 at 100 rpm (Fig. 5). It is interesting to highlight that the traditional USP Apparatus 1 utilizes a basket made by a screen with welded seam, with wire openings of 360 to 440 μm (10), compatible with the 420-μm initially used in USP Apparatus 3. This fact suggested that both the sieve mesh size and the Apparatus hydrodynamics play an important role in order to retain the drug product inside of the USP Apparatus 3 cylinders.
In order to minimize the passage of the powder through the sieve, an additional test was conducted with a 150-μm sieve and a dip rate of 5 dpm. Applying this condition, the dissolution profiles were similar, considering both f2 and DE values (p > 0.05) (Table V).
According to these findings, it is possible to conclude that USP Apparatus 3 provided similar dissolution profiles of hydrochlorothiazide 25 mg tablets compared to the compendial dissolution method using USP Apparatus 1, 100 rpm, when appropriate test conditions were applied.
To evaluate a higher dosage product, hydrochlorothiazide 50 mg tablets were tested according to the compendial method using USP Apparatus 1 and using USP Apparatus 3 at 5 dpm and a 150-μm sieve. The dissolution profiles of hydrochlorothiazide 50 mg tablets are represented in Fig. 6.

Dissolution profiles (mean ± SD) of hydrochlorothiazide 50 mg tablets
The compendial method and the proposed USP Apparatus 3 method reached a cumulative drug release of 101.7 ± 2.3% and 104.3 ± 1.9% at 60 min, respectively. As reported to hydrochlorothiazide 25 mg tablets, the dissolution profiles using the compendial and USP Apparatus 3 methods were similar (f2 = 51.8). Statistically similar DE values of 89.9 ± 1.3 and 92.3 ± 2.3 were obtained for USP Apparatus 1 and 3, respectively (p > 0.05). Therefore, the proposed USP Apparatus 3 method also provided a similar dissolution profile when compared to the compendial method to hydrochlorothiazide 50 mg tablets.
CONCLUSION
The dissolution testing is a quality control requirement and key tool to assess the performance of oral solid dosage forms. This test should simulate, as much as possible, the physiological conditions to which the drug product will be submitted in the gastrointestinal tract.
USP Apparatus 1 and 2 are the most used dissolution Apparatus in official monographs of the main pharmacopoeias from all over the world. Despite this fact, they have several disadvantages, being affected by shaft wobble, location, centering, and coning (5,7,8,29). The present study proposed the use of USP Apparatus 3 for dissolution testing of three antihypertensive IR drug products, which are typically analyzed using USP Apparatus 1 and 2.
It was demonstrated that USP Apparatus 3 could provide similar dissolution profiles when compared to the compendial methods using USP Apparatus 1 and 2 to the drug products tested when proper operational conditions are applied. In addition, the hydrodynamics of USP Apparatus 3 was capable to avoid the coning effect observed when using USP Apparatus 2 for atenolol tablets.
However, this study emphasized that USP Apparatus 3 methods for dissolution testing of IR tablets need to be specifically designed to each drug product under investigation. These methods should be developed considering all variables of the equipment (e.g., media volume, dip rate, and sieve mesh size) and characteristics of the drug and the drug product (e.g., solubility, BCS class, sink conditions, and dissolution specification), in order to achieve similar performance when compared to the other compendial Apparatus.
It is also important to highlight that analyses of failing batches of the drug products using the developed USP Apparatus 3 methods should be performed, aiming to evaluate their discriminative power. This evaluation will help to determine if the USP Apparatus 3 methods could be used as an alternative to the compendial methods, since the purpose of the dissolution testing is to discriminate between passing and failing batches, in a quality control perspective. As this work was designed to evaluate commercially available drug products, analyses of failing batches could not be performed. However, the results obtained provided good evidence of the possible use of the new developed methods using USP Apparatus 3 for dissolution testing of atenolol, captopril, and hydrochlorothiazide IR tablets.
References
Mwila C, Khamanga S, Walker R. Development and assessment of a USP Apparatus 3 dissolution test method for sustained-release nevirapine matrix tablets. Dissolut Technol. 2016;23(3):22–30. https://doi.org/10.14227/DT230316P22.
Cardot J-M, Beyssac E, Alric M. In vitro–in vivo correlation: importance of dissolution in IVIVC. Dissolut Technol. 2007;14(1):15–9. https://doi.org/10.14227/DT140107P15.
Hörter D, Dressman J. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 2001;46(1–3):75–87. https://doi.org/10.1016/S0169-409X(00)00130-7.
Shohin IE, Grebenkin DY, Malashenko EA, Stanishevskii YM, Ramenskaya GV. A brief review of the FDA dissolution methods database. Dissolut Technol 2016;23(3):6–10. https://doi.org/10.14227/DT230316P6.
Borst I, Ugwu S, Beckett AH. New and extended applications for USP drug release apparatus 3. Dissolut Technol. 1997;4(1):11–8. https://doi.org/10.14227/DT040197P11.
Pezzini BR, Issa MG, Duque MD, Ferraz HG. Applications of USP apparatus 3 in assessing the in vitro release of solid oral dosage forms. Braz J Pharm Sci. 2015;51(2):265–72. https://doi.org/10.1590/S1984-82502015000200003.
Yu LX, Wang JT, Hussain AS. Evaluation of USP apparatus 3 for dissolution testing of immediate-release products. AAPS PharmSci. 2002;4:1):1–5. https://doi.org/10.1208/ps040101.
Webster GK, Jackson JD, Bell RG. Poorly soluble drugs: dissolution and drug release. Pan Stanford; 2017. 686 p.
Crist GB. 2009 trends in small-volume dissolution apparatus for low-dose compounds. Dissolut Technol. 2009;16(1):19–22. https://doi.org/10.14227/DT160109P19.
Li J, Yang L, Ferguson SM, Hudson TJ, Watanabe S, Katsuma M, et al. In vitro evaluation of dissolution behavior for a colon-specific drug delivery system (CODES) in multi-pH media using United States Pharmacopeia apparatus II and III. AAPS PharmSciTech. 2002;3(4):E33–22. https://doi.org/10.14227/DT160109P19.
USP. THE UNITED STATES PHARMACOPEIA. 39th ed. USP39–NF34. Rockville: United States Pharmacopeial Convention; 2016. 8016 p.
Klein S, Rudolph MW, Dressman JB. Drug release characteristics of different mesalazine products using USP apparatus 3 to simulate passage through the GI tract. Dissolut Technol 2002;9(4):6–12. https://doi.org/10.14227/DT090402P6.
Cacace J, Reilly EE, Amann A. Comparison of the dissolution of metaxalone tablets (Skelaxin) usingUSP apparatus 2 and 3. AAPS PharmSciTech. 2004;5(1):29–31. https://doi.org/10.1208/pt050106.
Mercuri A, Fares R, Bresciani M, Fotaki N. An in vitro–in vivo correlation study for nifedipine immediate release capsules administered with water, alcoholic and non-alcoholic beverages: impact of in vitro dissolution media and hydrodynamics. Int J Pharm. 2016;499(1–2):330–42. https://doi.org/10.1016/j.ijpharm.2015.12.047.
Sanghvi PP, Nambiar JS, Shukla AJ, Collins CC. Comparison of three dissolution devices for evaluating drug release. Drug Dev Ind Pharm. 1994 Jan 20;20(6):961–80. https://doi.org/10.3109/03639049409038344.
Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20. https://doi.org/10.1023/A:1016212804288.
Elder D, Holm R. Aqueous solubility: simple predictive methods (in silico, in vitro and bio-relevant approaches). Int J Pharm. 2013;453(1):3–11. https://doi.org/10.1016/j.ijpharm.2012.10.041.
Nart V. DESENVOLVIMENTO E AVALIAÇÃO DE SISTEMAS MULTIPARTICULADOS NA FORMA DE MINI Florianópolis. Universidade Federal de Santa Catarina; 2015.
Weich A, de Oliveira DC, de Melo J, Goebel K, Rolim CMB. Validation of UV spectrophotometric and HPLC methods for quantitative determination of atenolol in pharmaceutical preparations. Lat Am J Pharm. 2007;26(5):765–70.
CDER/FDA. Guidance for Industry Dissolution Testing of Immediate. Vol. 4, Center for Drug Evaluation and Research. 1997.
CDER/FDA. Guidance for Industry, Waiver of in vivo bioavailability and bioequivalence studies for immediate release solid oral dosage forms based on a biopharmaceutics classification system. Center for Drug Evaluation and Research. 2015.
EMA. Note for guidance on the investigation of bioavailability and bioequivalence. Committee for proprietary medicinal products. 2001.
Swami R, Singh G, Bhasin P, Dureja H. In vitro dissolution profile comparison: a tool for biowaivers based on BCS. J Pharm Res. 2011;10(2):73–6. https://doi.org/10.18579/jpcrkc/2011/10/2/89014.
Ozturk N, Kaynak MS, Sahin S. Comparison of dissolution profiles of commercially available lamivudine tablets. Dissolut Technol. 2015;22(4):38–43. https://doi.org/10.14227/DT220415P38.
Anderson NH, Bauer M, Boussac N, Khan-Malek R, Munden P, Sardaro M. An evaluation of fit factors and dissolution efficiency for the comparison of in vitro dissolution profiles. J Pharm Biomed Anal. 1998;17(4–5):811–22. https://doi.org/10.1016/S0731-7085(98)00011-9.
Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–33. https://doi.org/10.1016/S0928-0987(01)00095-1.
Qiu Y, Chen Y, Zhang GGZ, Liu L, Porter W. Developing solid oral dosage forms: pharmaceutical theory & practice: Academic Press; 2009.
Kostewicz ES, Abrahamsson B, Brewster M, Brouwers J, Butler J, Carlert S, et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur J Pharm Sci. 2014;57:342–66. https://doi.org/10.1016/j.ejps.2013.08.024.
Seeger N, Lange S, Klein S. Impact of vibration and agitation speed on dissolution of USP prednisone tablets RS and various IR tablet formulations. AAPS PharmSciTech. 2015;16(4):759–66. https://doi.org/10.1208/s12249-015-0356-3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Guest Editor: Sandra Klein
Rights and permissions
About this article

Cite this article
Espíndola, B., Bortolon, F.F., Pinto, J.M.O. et al. New Approach for the Application of USP Apparatus 3 in Dissolution Tests: Case Studies of Three Antihypertensive Immediate-Release Tablets. AAPS PharmSciTech 19, 2866–2874 (2018). https://doi.org/10.1208/s12249-018-1086-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-018-1086-0