
Abstract
Rilpivrine is BCS class II drug used for treatment of HIV infection. The drug has low aqueous solubility (0.0166 mg/ml) and dissolution rate leading to low bioavailability (32%). Aim of this work was to enhance solubility and dissolution of rilpivirine using beta-cyclodextrin-based nanosponges. These nanosponges are biocompatible nanoporous particles having high loading capacity to form supramolecular inclusion and non-inclusion complexes with hydrophilic and lipophilic drugs for solubility enhancement. Beta-cyclodextrin was crosslinked with carbonyl diimidazole and pyromellitic dianhydride to prepare nanosponges. The nanosponges were loaded with rilpivirine by solvent evaporation method. Binary and ternary complexes of drug with β-CD, HP-β-CD, nanosponges, and tocopherol polyethylene glycol succinate were prepared and characterized by phase solubility, saturation solubility in different media, in vitro dissolution, and in vivo pharmacokinetics. Spectral analysis by Fourier transform infrared spectroscopy, powder X-ray diffraction, and differential scanning calorimetry was performed. Results obtained from spectral characterization confirmed inclusion complexation. Phase solubility studies indicated stable complex formation. Saturation solubility was found to be 10–13-folds higher with ternary complexes in distilled water and 12–14-fold higher in 0.1 N HCl. Solubility enhancement was evident in biorelevant media. Molecular modeling studies revealed possible mode of entrapment of rilpivirine within β-CD cavities. A 3-fold increase in dissolution with ternary complexes was observed. Animal studies revealed nearly 2-fold increase in oral bioavailability of rilpivirine. It was inferred that electronic interactions, hydrogen bonding, and van der Waals forces are involved in the supramolecular interactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
One of the major route of drug delivery is the oral route, especially for the treatment of many chronic diseases. High lipophilicity of 50% of the drugs, however, is a major deterrent for oral delivery. Biopharmaceutical classification system class II and IV drugs are a challenge to formulators, as their poor bioavailability is primarily caused by poor water solubility resulting in low drug absorption [1, 2]. To overcome these problems, various strategies have been adopted such as inclusion complexation, drug micronization, prodrug formation, and solid dispersions. Lipid-based drug delivery systems such as self-nanoemulsifying [3, 4] and self-microemulsifying drug delivery system (SNEDDS and SMEDDS) have also seen a surge in interest among formulators for their role in enhancing solubility and bioavailability [5].
Nanosponges (NS) are nanostructured carriers which have been prepared by reacting cyclodextrin with cross-linkers like diphenyl carbonate, pyromellitic dianhydride (PMDA), carbonyl diimidazole (CDI), and hexamethylene diisocynate [6, 7]. Beta cyclodextrin (β-CD)-based nanosponges (NS) are biocompatible and nanoporous carriers having the capability of forming supramolecular inclusion complexes as well as non-inclusion complexes with both hydrophilic and lipophilic drugs [8, 9]. The toxicological studies of the NS have proven them to be safe for use in pharmaceutical products. Shende et al. reported the toxicological safety of NS on the basis of acute and repeat dose toxicity studies on rats [10]. Extensive preclinical safety/toxicity assessments, in vitro cell line toxicity (evaluated on MCF-7, HT-29, Vero, HCPC-I cell lines), and hemolytic activity assessment have also been performed on NS [11].
Drug loading can be improved by varying the cyclodextrin (CD) and crosslinker ratio and to obtain a customized release profile. NS may be synthesized by either melt or solvent method [12, 13]. The formation of van der Waals, hydrophobic, and hydrogen bond interactions are causative factors in the complex formation as well as the removal of water molecules from the hydrophobic cavity [14].
NS have been used as carriers for enhancement of solubility and stability for drugs such as telmisartan [15, 16], meloxicam [17, 18], curcumin [19], efavirenz [20], cefpodoxime proxetil [21], acyclovir [22], and resveratrol [23]. They also have been used for taste masking of oseltamivir phosphate [24] and gabapentin [25]. A controlled release nanoparticle system containing NS have shown potential for delivery of antivirals, proteins, anti-inflammatory drugs, and for anticancer therapeutics which includes camptothecin, tamoxifen, quercetin, temozolomide, doxorubicin, and 5-fluorouracil [11, 26].
Non-nucleoside reverse transcriptase inhibitors (NNRTI) are usually combined with high activity antiretroviral therapy (HAART) as first-choice drugs to extend the lifespan of human immuno deficiency virus (HIV)-infected patients. NNRTIs block the reverse transcriptase enzyme thereby preventing uninfected cells from becoming infected. These include nevirapine, dolutegravir, efavirenz, rilpivirine, and etravirine [27].
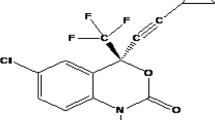
Rilpivirine (RPV) was selected for the study as it is a Biopharmaceutical Classification Scheme (BCS) class II drug having low aqueous solubility (0.0166 mg/ml) and high intestinal permeability. Its lipophilic nature is reflected in its log P of 3.8. RPV is 4-{[4-({4-[E-2-cynovinyl]-2,6-dimethylphenyl}amino) pyrimidin-2-yl]amino}benzonitrile (Fig. 1). It is used in the treatment of HIV infection as a second-generation NNRTI with better potency, longer half-life, and reduced side effects compared with older NNRTIs, such as efavirenz. RPV is accepted widely in the treatment of HIV infection, but its low bioavailability (32%) is attributed to poor solubility and slow dissolution rate which delays onset of action [28]. Kommavarpupu et al. prepared and characterized solid dispersions of RPV and Kollidon VA4 for enhancement of solubility and dissolution rate which showed a significant decrease in crystallinity of RPV, which remarkably increased its aqueous solubility and dissolution rate [29]. Vijay Kumar et al. reported enhanced solubility, dissolution rate, and oral bioavailability by developing self-emulsifying drug delivery system of RPV [30]. Ternary systems of itraconazole, NS, and copolyvidonum have been studied previously in which the auxiliary substance in conjugation with CD has been reported to improve the physicochemical, chemical, and bioavailability of guest molecule [31].

Molecular structure of rilpivirine
In the present work, binary complexes and ternary complexes of RPV with different carriers like β-CD, hydroxyl propyl (HP β-CD), NS, and tocopherol polyethylene glycol succinate (TPGS) were prepared. Two different β-CD-based NS crosslinked with pyromellitic dianhydride (PMDA) and carbonyl diimidazole (CDI) were prepared and their effect on solubility of RPV was studied by preparing binary and ternary complexes. TPGS was selected for ternary complex as auxiliary substance to study its effect on solubility of RPV in combination with NS. The complex formation was confirmed and characterized by predictive molecular modeling, Fourier transform infrared spectroscopy (FTIR), powder X-ray diffraction studies (PXRD), differential scanning calorimetry (DSC) and scanning electron microscopy (SEM), in vitro dissolution, and in vivo studies.
MATERIALS AND METHODS
Materials
Rilpivirine (RPV) was a gift sample from Mylan Pharmaceuticals, Hyderabad (India). The β-CD and HP β-CD were purchased from S. D. Fine Chemicals Ltd., Mumbai (India). Tocopherol polyethylene glycol succinate (TPGS) was gifted by Spectrochem Pvt. Ltd. Mumbai (India). Analytical grade reagents and solvents were used for the studies.
Methods
Synthesis of Carbonyl Diimidazole Nanosponges
A 250-ml-round bottom flask contained 25.00 g of anhydrous β-CD and 14.29 g of 1,1′-carbonyldiimidazole (CDI) solubilized in 150 ml of anhydrous N,N-dimethyl formamide (DMF). The solution was heated in an oil bath, at 90 °C until a gel was formed. After 24 h, a monolithic block was formed which was finely ground in a mortar, washed with deionized water in a Buchner funnel, in order to remove the solvent, the byproduct (imidazole) and possible unreacted monomers. Finally, the polymer was rinsed with ethanol and extracted in ethanol for approximately 45 h with Soxhlet extractor. The white powdery product was dried overnight at 60 °C in an oven and subsequently ground in mortar [32]. These NS are henceforth denoted as CDI-NS.
Synthesis of Pyromellitic Dianhydride Nanosponges
A 250-ml-round bottom flask containing 24.43 g of anhydrous β-CD solubilized in 100 ml of dimethylsulfoxide (DMSO) was placed in a water bath at room temperature. To this, 25 ml of triethylamine was added followed by 18.78 g of pyromellitic dianhydride. The solution was stirred until gelation point and a monolithic block were formed. The organogel obtained was finely powdered in a mortar, washed first with deionized water, and then followed by acetone and filtered through Buchner funnel. After drying, the polymer was extracted in acetone for approximately 45 h with Soxhlet extractor. The white product was dried overnight in an oven at 60 °C and followed by grinding in mortar [33, 34]. These NS are henceforth denoted as PMDA-NS.
Particle Size Reduction of NS
The NS were further subjected to particle size reduction to obtain nanosized products. A weighed amount of NS was suspended in saline solution (NaCl 0.9% w/v) at the concentration of 10 mg/ml under stirring at room temperature. The suspension was then dispersed using a high shear homogenizer (Ultraturrax®, IKA, Konigswinter, Germany) for 5 min at 24,000 rpm. The sample was subjected to 3 cycles of high pressure homogenization (HPH) for 90 min at a back-pressure of 500 bar, using an EmulsiFlex C5 instrument (Avastin, USA) to further reduce the size of the nanosponges and to obtain an almost homogenous nanoparticle distribution.
Evaluation of Plain NS
The particle size and polydispersity index of the NS were measured by Malvern Zeta Sizer (Model no: ZS90). The samples were diluted with DW prior to measurements. Zeta potential measurements were determined using an additional dip cell electrode in the same instrument. An average electric field of about 16.25 V/cm and an average current of about 0.05 mA were applied.
Quantitative Determination of RPV
High-performance liquid chromatography (HPLC) method was used for quantitative determination of RPV (Agilent Technologies; 1120 compact LC). The separation was performed using a C18 analytical column (4.6 × 250 mm). The mobile phase consisted of a combination of ortho-phosphoric acid and acetonitrile (50:50 v/v), filtered through a 0.45-μm membrane filter, and degassed using an ultrasonic bath before use. The mobile phase was introduced at a flow rate of 1.2 ml/min, the detection wavelength was 285 nm, and the injection volume was 15 μl. The HPLC method was validated for linearity and the method was found to be linear in the range of 62.5–375 ppm, respectively [35].
Phase Solubility Studies
Phase solubility studies for RPV with β-CD, HP β-CD, CDI-NS, and PMDA-NS were carried out by adding excess amount of drug to 20 ml of distilled water (DW) containing increasing concentrations of 5–25 mg β-CD or NS [36]. The contents were stirred using orbital shaker (Remi CIS-24BL) for 24 h at 25 ± 0.5 °C. The samples were then filtered using Whatman filter paper no. 41 and quantified by HPLC method (Agilent technologies; 1120 compact LC).
Preparation of Complexes
Binary and ternary complexes were prepared by solvent evaporation method [37,38,39,40]. In brief, RPV and CDI-NS (1:2 weight ratios) were dispersed in dichloromethane and triturated until the solvent evaporated. The moist solid dispersion was dried in an oven (Lab oven Biomedica) overnight at 50 °C to remove traces of dichloromethane. Other complexes were prepared using PMDA-NS, β-CD, and HP-β-CD all in weight ratios of 1:2 (Table I) using similar procedure. The ternary complexes of drug with NS and TPGS were prepared in weight ratio of 1:2:0.25. Saturation solubility studies and spectral analysis were carried out to characterize the complexes.
Evaluation and Characterization of Complexes
Saturation Solubility Studies
Saturation solubility studies were performed by adding excess drug/binary or ternary complexes to 20 ml of various media which included DW, 0.1 N HCl, phosphate buffer pH 6.8, fasting state simulated intestinal fluid (FaSSIF), and fed state simulated intestinal fluid (FeSSIF) separately and were equilibrated in an orbital shaker (Remi CIS-24BL) at 37 ± 0.5 °C for 24 h at 70 rpm [41, 42]. The suspensions were filtered through membrane filter (0.45 μm) and assayed for drug content of RPV by HPLC method. The studies were performed in triplicate.
Fourier Transform Infrared Spectroscopy
IR spectra of drug, binary, and ternary complexes were recorded using JASCO FTIR-460 Plus spectrophotometer to assess any interaction between RPV and NS and to confirm the formation of complexes. About 1–2 mg of samples was mixed with 50 mg dry potassium bromide and was subjected to transmission mode over the wave number range of 4000–400 cm−1.
Differential Scanning Calorimetry
Thermograms were obtained for RPV, binary, and ternary complexes using a MettlerToledoDSC823e differential scanning calorimeter. Heating rate of 5–10 °C/min was used and the temperature range was 30–300 °C. Standard aluminum sample pans were used; an empty pan was used as a reference material. Analysis was performed on 5-mg samples in nitrogen atmosphere (40 ml/min).
Powder X-Ray Diffraction
The PXRD spectra of RPV, plain NS, binary, and ternary complexes were recorded using high power powder X-ray diffractometer (Ru-200B) with Cu as target filter. A voltage/current of 40 kV/40 mA at a scan speed of 4θ/min was used. Analysis of the samples, i.e., RPV, NS, binary, and ternary NS complexes, was done at 2θ angle range of 5°–50°. Step time was 0.5 s and time of acquisition was 1 h.
Molecular Modeling Studies
Schrodinger Maestro software version 9.2 was used to construct the three-dimensional structures of RPV and β-CD and carry out molecular modeling studies to understand and decipher the mode of entrapment of drug molecule in the NS and β-CD.
In Vitro Dissolution Studies
Dissolution studies were performed for plain drug (25 mg) and complexes (equivalent to 25 mg RPV) in triplicate in 900 ml of phosphate buffer pH 6.8, using USP paddle type II dissolution apparatus at 37 ± 0.5 °C at 50 rpm. The plain drug and complexes were filled in hard gelatin capsules of size 3 for the studies. Samples were withdrawn at predecided intervals and analyzed using HPLC method at 285 nm. Cumulative percent release vs time (min) was plotted to obtain the dissolution profile.
Contact Angle Measurement
The contact angle (θ) of RPV, plain NS, binary, and ternary complexes was determined by using a micropipette to place 10 μl of water on surface of compacts of complexes. The compacts were prepared using hydraulic press (Technological search M-15) at 8–10 ton pressure. Photographs of the drop on surface of the compacts were taken after 10 s. It was traced on tracing paper and the contact angle was measured. Amaranth red was added to the water to ensure proper visibility of the drop [43].
In Vivo Pharmacokinetics Studies
Approval to carry out in vivo studies was obtained from Institutional Animal Ethics Committee, AISSMS College of Pharmacy (Approval from: AISSMS/CPCSEA/IAEC/PT-01/02/2K17) and their guidelines were followed for the studies. The in vivo performance of the complexes and RPV was evaluated in rats (Wistar albino rat).
Five groups (standard, test 1, test 2, test 3, and test 4) of six rats each weighing 200–250 g were fasted overnight before administration of the drug. Suspension of plain drug, binary, and ternary complexes (equivalent to 25 mg/kg) were prepared in DW. These suspensions were administered orally to rats through a feeding needle. Ether was used to anesthetize the rats for withdrawal of blood samples (0.5 ml) from the retro orbital vein at 0 (pre-dose), 0.5, 1, 2, 3, 4, and 5 h into tubes containing anti-coagulant, mixed and centrifuged at relative centrifugal force of 76 g for 30 min [44, 45]. Proteins were removed by precipitation by vortexing the supernatant with acetonitrile for 3 min and centrifuged as before for 10 min. The supernatant was then analyzed by bioanalytical RP-HPLC method (Agilent technologies; 1120 Compact LC) using C18 column with methanol/acetonitrile (90:10% v/v) as mobile phase at flow rate 2 ml/min. The detection wavelength (λmax) to quantify the amount of drug in plasma was set at 285 nm [46].
Pharmacokinetic Data Analysis
The pharmacokinetic parameters of binary and ternary complexes were calculated and compared with RPV. The time to reach maximum plasma concentration (tmax) and maximum plasma concentration (Cmax) were obtained from plasma concentration–time curve. Area under the concentration time curve (AUC0–5h) and elimination constant (Kel) were determined. Data was analyzed using one-way ANOVA using GraphPad Software Version 7.0.
RESULTS AND DISCUSSION
Evaluation of Plain NS
In this study, NS were synthesized using β-CD cross-linked with CDI and PMDA. Carbonate bonds are formed between β-CD molecules with CDI as the cross linker whereas PMDA forms ester link between β-CD molecules due to reaction between hydroxyl groups of β-CD and active carbonyl groups of CDI and PMDA (Figs. 2 and 3).

Schematic representation of formation of CDI crosslinked β-CD nanosponge [26]

Schematic representation of formation of PMDA crosslinked β-CD nanosponge [9]
The particle size of the NS was found to be 457 and 517 nm with polydispersity index of 0.23 and 0.19, respectively. Presence of NaCl during homogenization using high pressure homogenizer led to stabilization of the colloidal system. This caused reduction in particle size and polydispersity index. Zeta potentials were found to be − 20.2 mV for CDI-NS and − 21.55 mV for PMDA-NS. The high zeta potential is indicative of higher magnitude of repulsive forces which will reduce the tendency for particle aggregation.
Phase Solubility Studies
Phase solubility studies help in unraveling the stoichiometry and strength of interaction of inclusion complexation. As per the model suggested by Higuchi and Connors, the phase solubility diagrams revealed AL type curves of RPV with all complexing agents under study. This indicated formation of inclusion complex in 1:2 stoichiometric ratios. The slope of linear portion of phase solubility curve, indicative of stability constant (Kc), was calculated (Table II). The Kc values above 150 M−1 indicate good complexation ability [47]. All the complexes displayed reasonably strong complexation strength with the PMDA-NS showing the highest strength of interaction with RPV.
Evaluation and Characterization of Complexes
Saturation Solubility Studies
Saturation solubility studies of RPV in various complexes revealed enhanced solubility in presence of various carriers. Both NS in the form of binary complexes with RPV showed superior solubilization effect in all media. A 10- to 13-fold increase was evident in DW, while in 0.1 N HCl, the increase was to the tune of 12- to 14-fold in CDI-NS and PMDA-NS, respectively. Ternary complexes, with TPGS as the auxiliary ingredient, showed a further increase in solubility of RPV in all media. A significant increase in solubility was observed in phosphate buffer pH 6.8 for both binary and ternary complexes with both NS. The PMDA-NS was found to have a greater effect on solubility of RPV than CDI-NS. Ternary NS complexes displayed ~ 12-fold increase in FaSSIF and ~ 10-fold increase in FeSSIF (Fig. 4). An insignificant increase in solubility was evident with binary and ternary complexes of β-CD and HP-β-CD. RPV, being moderately lipophilic (log P = 3.8), may be easily entrapped within the hydrophobic toroidal cavity of β-CD and HP β-CD. However, the molecular size and conformation of the guest molecule are critical determinants of the inclusion complexation. Molecular modeling studies using Schrodinger maestro software version 9.2 (Fig. 5) revealed that the length of the RPV molecule is 17.2 Å while its width, measured across the center, is 7.9 Å. β-CD molecule resembles a truncated cone with an internal diameter of 6–6.5 Å and height of the cone is approximately 7.9 Å [7, 20]. Thus, we may infer that RPV is partially entrapped within the β-CD cavities. RPV molecule consists of three primary functional groups, i.e., cyanovinyl group, pyrimidine group, and benzonitrile ring. The pyrimidine group is more lipophilic than the other groups, and hence, this group may be getting entrapped within the hydrophobic β-CD cavity [48, 49]. The molecular modeling images reveal the possible mode of entrapment of RPV within the β-CD cavities. It appears that the RPV molecule is straddled by β-CD with the benzonitrile and cyanovinyl groups projecting outwards. The CDI-NS are β-CD molecules crosslinked with carbonyl groups. Greater enhancement in solubility of RPV in binary complexes with CDI-NS is observed. This may be attributed to supplementary interactions such as van der Waals forces and hydrogen bonding between the amino groups of RPV and the carbonyl group [50]. The PMDA-NS are acid NS present almost completely in the salt form. Hence, the cross link sites are capable of cation exchange reactions [51]. RPV is a basic drug with pKa of 12.9 and 5.6 [52]. The cyanovinyl group of RPV is basic in nature and we may presume that it will undergo electrostatic interactions with the carboxyl group of PMDA-NS. Similar interactions have been reported for acyclovir-loaded carboxylated nanosponges [22]. Hydrophobic interactions, dipole-dipole, and electrostatic interactions have been reported between complexing agent and guest molecule in inclusion complexation [53].

Saturation solubility studies of complexes in various media (n = 3, mean ± SD)

Molecular modeling using Schrodinger Maestro software. a Molecular dimensions of RPV, b–d possible mode of entrapment of RPV in β-CD
Presence of TPGS in the ternary complexes was found to produce a modicum of increase in RPV solubility. TPGS is a polymeric surfactant and is widely reported as a solubilizer and wetting agent for drugs like resveratrol [54]. In the present study, it may be enhancing the interaction between the NS and RPV by reducing the interfacial tension between drug and NS. Besides this, the critical micelle concentration (CMC) of TPGS is reported to be 0.02% w/v [55] and the concentration of TPGS in the dissolution media exceeds its CMC. Thus, greater enhancement in solubility in ternary complexes of both NS may be due to auxiliary micellar solubilization. The solubility was enhanced over 13-fold and 12-fold in case of ternary complexes with both NS in FaSSIF and FeSSIF. RPV itself showed higher solubility in FaSSIF and FeSSIF. The soya lecithin and sodium taurocholate in the two media may themselves be forming simple and mixed micelles with TPGS. Hence, a cumulative increase in RPV solubility is evident in FaSSIF and FeSSIF.
Fourier Transform Infrared Spectroscopy
The IR spectra of RPV, binary, and ternary complexes were recorded to corroborate findings (Fig. 6). The characteristic FTIR spectra of RPV show C ≡ N stretch at 2216.58 cm−1. C–N stretch at 1198 cm−1, C–H bend at 757.75 cm−1, C=C stretch at 1629.48 cm−1, and N–H bend at 1594.18 cm−1. The FTIR spectra of NS showed a peak at 1649 cm−1 which is typical of the ester group. The complexation between RPV and NS was confirmed by FTIR studies. IR spectra of complex showed that the characteristic peaks seen in spectra of plains NS and RPV were shifted. The drug-excipient compatibility studies revealed that RPV and both NS are compatible [56].

FTIR spectra of RPV and inclusion complexes
Differential Scanning Calorimetry
DSC thermogram of RPV showed sharp endothermic peak at 243 °C which reflects the melting point (Fig. 7a, b). Minimal enthalpy changes occurred during the melting process as reflected in the ΔH value of 1.21 mJ. The disappearance of the endotherm in the ternary complexes indicated that RPV is dispersed in the molecular form in the NS [13, 57]. The thermogram of binary and ternary complexes displayed shallow and broadened peaks at approximately 120–130 °C. The inference that can be drawn from these thermograms is that the drug is molecularly dispersed in the NS with lipophilic portions of RPV being entrapped within the hydrophobic cavities [58].

a, b DSC thermograms of RPV and inclusion complexes
Powder X- Ray Diffraction
XRD spectra for RPV, CDI-NS, PMDA-NS, binary, and ternary complexes further provided evidence of complexation. Diffraction pattern of RPV showed numerous distinctive peaks at 2θ angles of 21°, 22°, 24°, 29°, 32° throughout the scanning range which indicated its highly crystalline nature (Fig. 8). Formation of inclusion complexes was confirmed by the reduced peak intensities and disappearance of some peaks in spectra of binary and ternary complexes with both CDI-NS and PMDA-NS. The crystallinity of RPV was considerably reduced in the ternary complexes as compared to binary NS complexes. TPGS, being a polymeric surfactant, may be interfering with the crystallization process during complexation, and hence, a greater reduction in peak intensities is evident. Similar findings have been reported by Martins et al., who linked reduced peak intensity in X-ray diffractogram to inclusion complexation [59]. Janet et al. also reported formation of partial crystallinity in inclusion complexes of beta-sitosterol [60]. We may conclude from their findings that decrease in peak intensities in X-ray spectra and broadening of peak in thermogram is indicative of inclusion complexation. The procedure may lead to partial amorphization of guest molecules which get entrapped into the CD cavities. Presence of the complexing agent may be interfering with the crystallization process and hence leading to partial amorphization of the drug. Amorphization is reported as a method for solubility enhancement of poorly soluble drugs. However, pure amorphous drugs are rarely used in formulation development because of their innate physical and/or chemical instability. Hence, additives such as polymers are used to stabilize the amorphous form of drugs. The amorphous drug gets entrapped into the cross-linked networks of polymers. This obstructs the ability of the molecules to move and thereby preserves the solubility and stability of the amorphous state throughout the shelf life of the product [61, 62]. Thus, we may infer that NS not only facilitate inclusion complexation of the drug but also accord stability to the amorphous RPV.

PXRD spectra of RPV and inclusion complexes
In Vitro Dissolution Studies
In vitro dissolution studies were carried out for binary and ternary complexes in phosphate buffer pH 6.8 (Fig. 9). Binary complexes showed a release of ~ 56 to 68% in CDI-NS and PMDA-NS, respectively, in 60 min. Ternary complex of NS exhibited significantly higher release of 81 to 92%, respectively, in the same span of time. The percent cumulative drug release was increased about three-fold for ternary complex and about two-folds for binary complexes compared with RPV. Increase in dissolution can be correlated with enhancement in saturation solubility. Increase in dissolution might be due to entrapment of RPV molecules in hydrophobic cavity of β-CD in the NS structure and also increase in wettability due to addition of TPGS. TPGS is a hydrophilic protective polymeric surfactant [63], could be enhancing the wettability of free RPV particles in case of ternary complex. The TPGS could also be solubilizing the RPV molecules which are not entrapped in NS through micellar solubilization. This synergistic effect of solubility relates well with enhancement of dissolution in case of ternary complex. RPV is a lipophilic drug with log P of 3.8. To confirm the above rationale, the effect of TPGS and NS on the wettability of RPV was measured by determining contact angle of water drop on compacts. The contact angles for compacts of plain RPV, binary NS complexes (IC3 and IC4), and ternary NS complexes (IC7 and IC 8) are reported in Table III. Decrease in contact angle (θ) is indicative of increased wettability [64]. A significant decrease in contact angle was evident in compacts prepared with ternary complexes (i.e., IC7 and IC8). This could be primarily due to the hydrophilic nature of TPGS and its capacity to reduce solid-liquid interfacial tension [65, 66]. The high contact angle of RPV can be attributed to its hydrophobic nature (log P = 3.8). The compacts prepared with plain NS displayed better wettability than plain drug as reflected by their lower contact angles (Table III). This is an obvious finding as both NS have hydrophilic character imparted to due to the crosslinking. The compacts prepared with binary complexes of both NS (IC3 and IC4) also showed lower contact angles. Rao et al. reported a decrease in contact angle of binary NS complex of efavirenz as compared to the free drug and attributed it to presence of number of hydroxyl functional groups projecting from the external surfaces of the NS similar to that of β-CD which interact with water molecules by hydrogen bonding [20].

In vitro dissolution profile of RPV and inclusion complexes in phosphate buffer pH 6.8 (n = 3, mean ± SD)
In Vivo Pharmacokinetic Studies
The ternary and binary complexes which showed a marked enhancement in saturation solubility and dissolution rate were subjected to pharmacokinetic profiling. These complexes were compared with RPV. The Cmax for RPV, binary, and ternary complexes were found to be 2.192 ± 0.112, 3.025 ± 0.23, 4.032 ± 0.71 μg/ml and tmax were 4, 3, 2 h, respectively (Table IV). The relative bioavailability of RPV was enhanced nearly 2–3-folds for ternary complexes (IC7 and IC8), compared to RPV (Fig. 10). The increase in solubility and dissolution rate can explain the increased bioavailability. Graphpad prism7 software was used to statistically validate the results. Analysis of variance (ANOVA) test was used to detect differences in five groups. The probability p value was below 0.05 and this indicated that there is appreciable difference in pharmacokinetics of the formulations. A moderate increase in the elimination rate constant (Kel) was seen for binary and ternary complexes of both NS. It is reported that hepatic metabolism is primarily responsible for elimination of RPV [67] and also that 25% of dose of unchanged RPV was excreted via feces [68]. Drug absorption in the gastrointestinal tract is a complex process involving efflux pumps, influx solute carrier transporters, intracellular metabolism, passive diffusion that depends on the physicochemical properties of the drug and the interactions between these parameters. Anti retroviral drugs are reported to be absorbed by carrier mediated transport mechanism which are saturable in nature and also are known to degrade in the gastric pH [69]. The NS provide protection to RPV from the aggressive gastric environment as also control the release of RPV. Besides this, poorly water soluble drugs when encapsulated in nanocarriers are transported through the Peyer’s patches, followed by, entry into the intestinal lymphatic transport via the Mcells and eventually into the systemic circulation [70]. Thus, endocytosis and phagocytosis mechanisms involved in this transport model minimize the hepatic first-pass metabolism of therapeutic agents, therefore improving their bioavailability [71]. In lieu of this, we may presume that RPV encapsulated in NS are transported across the gastric mucosa by the aforementioned mechanism resulting in higher AUC. A moderate increase in elimination rate constant is seen. However, this may not have an adverse effect on pharmacological activity of RPV in the present study.

Plasma concentration time profile of RPV and inclusion complexes (n = 3, mean ± SD) (oral administration to rats at 25-mg/kg dose strength)
CONCLUSION
Cyclodextrins are capable of forming inclusion complexes with various molecules resulting in increased solubility of drugs. However, this approach is less effective for hydrophilic or medium polar drugs. Thus, nanosponges present a potential strategy to improve this drawback. Nanosponges are biocompatible cross-linked cyclodextrin polymers with the ability to encapsulate diverse molecules. In the present study, inclusion complexes of rilpivirine and β-cyclodextrin-based nanosponges, i.e., binary and ternary complexes, were developed. A remarkable increase in the solubility and dissolution rate of rilpivirine was evident. Consequently, an increase in bioavailability of rilpivirine was seen for binary and ternary complexes of both nanosponges.
References
Neslihan G, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drug. Biomed Pharmacotherapy. 2004;58:173–82.
Enin A, Hadel A. Self-nanoemulsifying drug-delivery system for improved oral bioavailability of rosuvastatin using natural oil antihyperlipidemic. Drug Dev Ind Pharm. 2014;41(7):1047–56.
Gao P, Guyton M, Huang T, Bauer J, Stefanski K. Enhanced oral bioavailability of a poorly water soluble drug by supersaturatable formulations. Drug Dev Ind Pharm. 2004;30(2):221–9.
Zhu J, Tang D, Feng L. Development of self-microemulsifying drug delivery system for oral bioavailability enhancement of berberine hydrochloride. Drug Dev Ind Pharm. 2013;39(3):499–506.
Peri D, Cleary R, Wyandt M, Jones A. Inclusion complexes of tolnaftate with β-cyclodextrin and hydroxypropyl β-cyclodextrin. Drug Dev Ind Pharm. 2008;20(8):1401–10.
Cavalli R, Trotta F, Tumiatti W. Cyclodextrin-based nanosponge for drug delivery. J Inclusion Phenom Macrocyclic Chem. 2006;56:209–13.
Gursalkar T, Bajaj A, Jain D. Cyclodextrin based nanosponges for pharmaceutical use. Acta Pharm. 2013;63:335–58.
Holvoet C, Vander Y, Plaizier J. Inclusion complexation of lorazepam with different cyclodextrins suitable for parenteral use. Drug Dev Ind Pharm. 2005;31(6):567–75.
Sanghavi M, Choudhari K, Matharu R, Viswanathan L. Inclusion complexation of lorazepam with β-cyclodextrin. Drug Dev Ind Pharm. 1993;19(6):701–12.
Pravin S, Yogesh K, Gaud RS, Kiran D, Roberta C, Francesco T, et al. Acute and repeated dose toxicity studies of different β-cyclodextrin-based nanosponge formulations. J Pharm Sci. 2015;104:1856–63.
Swaminathan S, Racalli R, Trotta F. Cyclodextrin-based nanosponges: a versatile platform for cancer nanotherapeutics development. WIREs Nanomed Nanobiotechnol. 2016;8:579–601.
Ahmed A, Patil G, Zaheer Z. Nanosponges—a completely new nano-horizon: pharmaceutical applications and recent advances. Drug Dev Ind Pharm. 2013;39(9):1263–72.
Christian F, Mehrdad Y, Claudio O. Inclusion and functionalization of polymers with cyclodextrins: current applications and future prospects. Molecules. 2014;19:1466–79.
Gidwani B, Vyas A. A comprehensive review on cyclodextrin based carriers for delivery of chemotherapeutic cytotoxic anticancer drugs. Bio Med Res Int. 2015;15:1–15.
Rao M, Bajaj A, Khole I, Munjapara G, Trotta F. In vitro and in vivo evaluation of β-cyclodextrin based nanosponges of telmisartan. J Inclusion Phenom Macrocyclic Chem. 2013;77(4):135–45.
Zhong L, Zhu X, Yu B, Su W. Influence of alkalizers on dissolution properties of telmisartan in solid dispersions prepared by co-grinding. Drug Dev Ind Pharm. 2014;40:1660–9.
Shende P, Gaud R, Bakal R, Patil D. Effect of inclusion complexation of meloxicam with betacyclodextrin-and beta-cyclodextrin-based nanosponges on solubility, in vitro release and stability studies. Colloids Surf, B. 2015;15:1–26.
El-Badry M, Fathy M. Enhancement of the dissolution and permeation rates of meloxicam by formation of its freeze-dried solid dispersions in polyvinyl pyrrolidone K-30. Drug Dev Ind Pharm. 2006;32:141–50.
Darandale S, Vavia P. Cyclodextrin based nanosponges of curcumin: formulation and physiochemical characterization. J Inclusion Phenom Macrocyclic Chem. 2013;75:315–22.
Rao M, Shirsath C. Enhancement of bioavailability of non-nucleoside reverse transcriptase inhibitor using nanosponges. AAPS PharmSciTech. 2016;10:1–11.
Rao M, Bajaj A, Pardeshi A, Aghav S. Investigation of nanoporous colloidal carrier for solubility enhancement of Cefpodoxime proxetil. J Pharm Res. 2012;5(5):2496–9.
Lemboa D, Swaminathan S, Donalisioa M, Trotta F, Cavalli R. Encapsulation of acyclovir in new carboxylated cyclodextrin-based nanosponges improves the agent’s antiviral efficacy. Int J Pharm. 2013;443:262–72.
Ansari K, Vavia P, Trotta F, Cavalli R. Cyclodextrin-based nanosponges for delivery of resveratrol: in vitro characterisation, stability, cytotoxicity and permeation study. AAPS PharmSciTech. 2011;12:132–46.
Sevukaranjan M, Bachala T, Nair R. Novel inclusion complex of oseltamivir phosphate with beta cyclodextrin physic-chemical characterization. J Pharma Sci Res. 2010;2(9):583–9.
Rao M, Bhingole R. Nanosponge based pediatric-controlled release dry suspension of gabapentin for reconstitution. Drug Dev Ind Pharm. 2015;41(12):2029–36.
Safwat M, Soliman G, Sayed D, Attia M. Gold nanoparticles capped with benzalkonium chloride and poly (ethylene imine) for enhanced loading and skin permeability of 5-fluorouracil. Drug Dev Ind Pharm. 2017;(11):1–16.
Hayashida T, Hachiya A, Ode H. Rilpivirine resistance mutation in HIV-1 reverse transcriptase predisposed by prevalent polymorphic mutations. J Antimicrob Chemother. 2016;71(10):2760–6.
Sharma P, Garg S. Pure drug and polymer based nanotechnologies for the improved solubility, stability, bioavailability and targeting of anti-HV drugs. Adv Drug Deliv Rev. 2010;62:491–502.
Kommavarpupu M, Sunkara M. Preparation and characterization of rilpivirine solid dispersions with the application of enhanced solubility and dissolution rate. J Basic Appl Sci. 2015;4:71–9.
Kumar V, Raja J, Bhikshapathi D. Enhancement of solubility and oral bioavailability of poorly soluble drug Rilpivirine by novel self-emulsifying drug delivery system. Am J Pharma Tech Res. 2015;5(3):1–18.
Swaminathan S, Vavia P, Trotta F. Formulation of beta cyclodextrin based nanosponges of itraconazole. J Inclusion Phenom Macrocyclic Chem. 2007;57:89–94.
Dora CP, Trotta F, Kushwah V, Devasari N, Singh C, Suresh S, et al. Potential of erlotinib cyclodextrin nanosponge complex to enhance solubility, dissolution rate, in vitro cytotoxicity and oral bioavailability. Carbohydr Polym. 2015;5:1–19.
Shankar G, Agrawal Y. Formulation and evaluation of β-cyclodextrin based nanosponges of a poorly water soluble drug. J Chem Pharm Res. 2015;7(4):595–604.
Trotta F, Shende P, Biasizzo M. Influence of different techniques on formulation and comparative characterization of inclusion complexes of ASA with b-cyclodextrin and inclusion complexes of ASA with PMDA cross-linked b-cyclodextrin. J Inclusion Phenom Macrocyclic Chem. 2015;3:14–27.
Ghosh S, Bomma S, Prasanna L, Vidyadhar S. Method development and validation of Rilpivirine in bulk and tablet doses form by RP-HPLC method. Res J PharmTech. 2013;6(3):1–8.
Torne S, Ansari K, Vavia P, Trotta F, Cavalli R. Enhanced oral paclitaxel bioavailability after administration of paclitaxel loaded nanosponges. Drug Deliv. 2010;17(6):419–25.
Andreza R, Ana F, Delfim S, Francisco V. Preparation and solid state characterization of inclusion complexes formed between miconazole and methyl-β-cyclodextrin. AAPS PharmSciTech. 2008;9(4):1102–9.
Darandale S, Shevalkar G, Vavia P. Effect of lipid composition in propofol formulations: decisive component in reducing the free propofol content and improving pharmacodynamic profiles. AAPS PharmSciTech. 2016:1–10.
Bilensoy E, Rouf M, Vural I, Sen M, Hincal A. Mucoadhesive, thermosensitive, prolonged-release vaginal gel for clotrimazole: β-cyclodextrin complex. AAPS Pharm SciTech. 2006;7(2):54–62.
Manca M, Marco Z, et al. Diclofenac-β-cyclodextrin binary systems: physicochemical characterization and in vitro dissolution and diffusion studies. AAPS Pharm SciTech. 2005;6(3):464–72.
Panikumar A, Venkat R, Sunitha G, Babu S, Subrahmanyam C. Development of biorelevant and discriminating method for dissolution of efavirenz and its formulations. Asian J Pharm Clin Res. 2012;5(3):220–3.
Bajaj A, Rao M, Pardeshi A, Sal D. Nanocrystallization by evaporative antisolvent technique for solubility and bioavailability enhancement of telmisartan. AAPS Pharm SciTech. 2012;13:1331–40.
Rao M, Shivpuje S, Godbole R, Shirsath C. Design and evaluation of sustained release matrix tablets using sintered technique. Int J Pharm Sci. 2015;8(2):115–21.
Date A, Shibata A, Bruck P, Christopher J. Development and validation of a simple and isocratic reversed-phase HPLC method for the determination of rilpivirine from tablets, nanoparticles and HeLacell lysates. Biomed Chromatogr. 2015;29(5):709–15.
Cozzia V, Charbe N, Baldelli M, Castoldia S. Development and validation of a chromatographic UV method for the simultaneous quantification of dolutegravir and rilpivirine in human plasma. Drug Monit. 2016:1–8.
Castold D, Clementib E, Cozzia V. Development of an HPLC–UV assay method for the simultaneous quantification of nine antiretroviral agents in the plasma of HIV-infected patients. J Pharm Anal. 2016;6:396–403.
Singireddy A, Subramanian S. Fabrication of cyclodextrin nanosponges for quercetin delivery: physicochemical characterization, photostability and antioxidant effects. J Mater Sci. 2014;49:8140–53.
Prasasty V, Yulandi A. Structure based design of novel rilpivirine analogues as HIV-1 non- nucleoside reverse transcriptase inhibitors through QSPR and molecular docking. Int J Pharm Sci. 2015;7(11):340–5.
Sagawa N, Shikata T. All are polar molecules hydrophilic? Hydration numbers of nitro compounds and nitriles in aqueous solution. Phys Chem Chem Phy. 2014;16(26):1–18.
Sanfrutos J, Jaramillo F, Mahmoud A. Divinyl sulfone cross-linked cyclodextrin based polymeric materials: synthesis and applications as sorbents and encapsulating agent. Molecules. 2015;20:3565–81.
Trotta F, Zanetti M, Cavalli R. Cyclodextrin-based nanosponges as drug carriers. Beilstein J Org Chem. 2012;8:2091–9.
Usach I, Melis V, Peris J. Non-nucleoside reverse transcriptase inhibitors: a review on pharmacokinetics, pharmacodynamics, safety and tolerability. J Int AIDS Soc. 2013;16(1):18567–77.
Sherje A, Dravyakar B, Kadam D, Jadhav M. Cyclodextrin-based nanosponges: a critical review. Carbohydr Polym. 2017;17:1–45.
Shengpeng W, Ruie C, Joe M, Michel R, Meiwan C. mPEG-b-PCL/TPGS mixed micelles for delivery resveratrol in overcoming resistant breast cancer. Expert Opin Drug Deliv. 2015;12(3):361–73.
Sharma N, Madan P, Lin S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: a cosurfactant study. As J Pharm Sci. 2016;11(3):404–16.
Takahashi A, Veiga F, Ferraz H. Cyclodextrins inclusion complexes characterization—part II: X-ray diffraction, infrared spectroscopy and nuclear magnetic resonance. Int J Pharm Sci Rev Res. 2012;12(1):9–15.
Kumar V, Raju J. Solubility enhancement of poorly soluble antiretrovirals by novel self-emulsifying drug delivery system. Int J Pharm Anal Res. 2016;5(2):245–56.
Shrinivas P, Sreeja K. Formulation and evaluation of voriconazole loaded nanosponges for oral and topical delivery. Int J Drug Dev Res. 2013;5(1):55–69.
Martins M, Calderini F. Host–guest interactions between dapsone and β-cyclodextrin (part II): thermal analysis, spectroscopic characterization, and solubility studies. J Inclusion Phenom Macrocyclic Chem. 2012;74(1):109–16.
Janet C, Olatunji A, Godwin A, Xia W, Khan I. Preparation and characterization of β sitosterol/ β-cyclodextrin crystalline inclusion complexes. J Inclusion Phenom Macrocyclic Chem. 2015;83(1):141–8.
Baghel S, Cathcart H, O’Reilly NJ. Polymeric amorphous solid dispersions: a review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J Pharm Sci. 2016;105:2527–44.
Orsolya J’j’r-L, Piroska S´-R’v’s. Amorphization of a crystalline active pharmaceutical ingredient and thermoanalytical measurements on this glassy form. J Therm Anal Calorim. 2010;102:243–7.
James MR. Influence of vitamin E TPGS on the properties of hydrophilic films produced by hot melt extrusion. Int J Pharm. 2000;202(2):63–70.
Yaun Y, Lee R. Contact angle and wetting properties. Surf Sci Tec. 2013;51:3–33.
Srivalli K, Mishra B. Improved aqueous solubility and antihypercholesterolemic activity of ezetimibe on formulating with HP-β-Cyclodextrin and hydrophilic auxiliary substances. AAPS PharmSciTech. 2016;17(2):272–83.
Thakkar H, Desai J. Influence of excipient on drug absorption via modulation of intestinal transporters activity. As J Pharm. 2015;9:69–82.
www.jaNSen.com/newzealand/sites/www_jaNSen_com.../edurant_data_sheet.pd (Date of access 13.05.17).
Lade J, Avery L, Bumpus N. Human biotransformation of the non-nucleoside reverse transcriptase inhibitor rilpivirine and a cross-species metabolism comparison. Antimicrob Agents Chemother. 2013;57(10):5067–79.
Sosnik A, Augustine R. Challenges in oral drug delivery of antiretrovirals and the innovative strategies to overcome them. Adv Drug Deliv Rev. 2016;1-44. https://doi.org/10.1016/j.addr.2015.12.022:105–20.
Florence A. The oral absorption of micro-and nanoparticulates: neither exceptional nor unusual. Pharm Res. 1997;14:259–66.
Desai M, Labhasetwar V, Amidon G, Levy R. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm Res. 1996;13:1838–45.
Acknowledgements
The authors would like to thank Dr. Ashwini Madgulkar Principal, AISSMS College of Pharmacy, Pune-01, Maharashtra (India) for providing necessary help and facilities to carry out the research work and also thankful to Mylan Laboratories, Hyderabad (India) for providing drug as gift sample.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Approval to carry out in vivo studies was obtained from Institutional Animal Ethics Committee, AISSMS College of Pharmacy (Approval from: AISSMS/CPCSEA/IAEC/PT-01/02/2K17) and their guidelines were followed for the studies.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Rao, M.R.P., Chaudhari, J., Trotta, F. et al. Investigation of Cyclodextrin-Based Nanosponges for Solubility and Bioavailability Enhancement of Rilpivirine. AAPS PharmSciTech 19, 2358–2369 (2018). https://doi.org/10.1208/s12249-018-1064-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-018-1064-6