
Abstract
Qualitative risk assessment methods are often used as the first step to determining design space boundaries; however, quantitative assessments of risk with respect to the design space, i.e., calculating the probability of failure for a given severity, are needed to fully characterize design space boundaries. Quantitative risk assessment methods in design and operational spaces are a significant aid to evaluating proposed design space boundaries. The goal of this paper is to demonstrate a relatively simple strategy for design space definition using a simplified Bayesian Monte Carlo simulation. This paper builds on a previous paper that used failure mode and effects analysis (FMEA) qualitative risk assessment and Plackett-Burman design of experiments to identity the critical quality attributes. The results show that the sequential use of qualitative and quantitative risk assessments can focus the design of experiments on a reduced set of critical material and process parameters that determine a robust design space under conditions of limited laboratory experimentation. This approach provides a strategy by which the degree of risk associated with each known parameter can be calculated and allocates resources in a manner that manages risk to an acceptable level.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
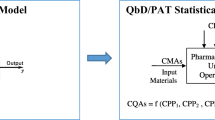
The regulatory framework outlined in the ICH guidance documents Q8 (R2) Pharmaceutical Development, ICH Q9 Quality Risk Management, and ICH Q10 Pharmaceutical Quality Systems was introduced to facilitate drug development using the quality-by-design (QbD) paradigm (1–3). The principal steps for the development of a new drug using QbD are shown in Fig. 1. In a previous study, the authors examined the initial steps of QbD for ciprofloxacin tablets. The present investigation uses a combination of process modeling with Monte Carlo simulation to determine a design space based upon risk analysis (4), see Fig. 1. The risk assessment begins with identification of the critical quality attributes (CQAs) which, if not achieved or maintained, represent the most severe risk outcomes. Figure 1 shows that this study continues the risk assessment focusing on the probabilities of harm represented by not meeting design CQAs. The flowchart in Fig. 1 also shows the overlap of design space principle with risk control concepts; given that design space definition and optimization suggest important process risk control strategies. The integration of the previous study with this research is discussed in the “RESULTS AND DISCUSSION” section.

QbD drug product development flow chart showing principal steps
The QbD paradigm of drug development may include describing a design space, which involves finding the parameter ranges for all CQAs that predict the product will meet the quality target product profile (QTPP). The ICH quality guidelines call for defining the design space under quality risk management (QRM) principles. QRM is growing rapidly in both theory and application to pharmaceutical product life cycle management (1,2,5,6), and an increasing number of pharmaceutical development teams are applying quantitative risk management approaches to pharmaceutical QbD (7–9). Qualitative risk management tools excel for building structural and quantitative models as support for a risk-based selection of critical quality attributes necessary for creating a design space. Quantitative tools for risk management provide risk-based statistical support for decisions about critical quality attributes and optimal formulation and process parameters and are needed for linking quality to public health consequences. Given the challenges inherent in directly measuring risks to patients, quality attributes often serve as surrogate measures in quality risk management. Although quantitative approaches to optimizing design space parameters are not new, the recent QbD efforts are novel applications of quality risk management as the framework for “risk-based thinking” when developing a design space or process quality systems, and these methods also extend the qualitative methods commonly practiced in the pharmaceutical industry.
Analysis of the uncertainties and risks have been applied to engineering design space problems for many years, perhaps notably beginning with the confidence interval methods of Box and Hunter (10). The analytical response surface methods of Box and others have experienced growing use and acceptance as screening tools for mapping design spaces (11–14). Popularity of the methods derives from the experimental efficiency attained in assuming models of smooth multivariate responses between design extrema. Probabilistic methods generally seek a mapping of the uncertainties among the variables and their interaction as a solution to finding optimal regions. The two approaches can be used in a complementary manner and both might be interpreted broadly in terms of risk.
The first major probabilistic risk analysis for a highly complex design problem is usually attributed to the nuclear Reactor Safety Study in 1975 (15). At the time of that seminal work and for the decades following, probabilistic risk simulations of design space problems, i.e., using Monte Carlo sampling, typically required access to mainframe computers and significant computation times. More recently, the rapid evolution of desktop computing power has brought Monte Carlo simulation into the realm of routine risk analysis and as such, probabilistic risk analysis using Monte Carlo methods support risk modeling for a wide range of disciplines (16,17) including design of experiment (DOE)-based process design (9,18,19). Numerous desktop software applications are now available as add-ons to spreadsheet software, components of sophisticated statistical packages, or as stand-alone software applications.
The QbD paradigm of product development requires an in-depth process understanding that can be challenging to achieve, given that there are potentially thousands of different combinations of process parameters that might affect the quality of the manufactured product (4). The problem is made worse by the fact that multivariate predictive models for pharmaceutical operations cannot be readily derived from first principles of physics and chemistry. Thus, most of our knowledge of optimal unit operations is based upon empirical methods followed by statistical inference to find the optimal process parameters. Developers of a design space and control strategy encounter the dilemma that studying too many variables will increase development costs and, perhaps, delay bringing a product to the market which can deprive patients of new, potentially lifesaving medicines. On the other hand, studying too few variables amounts to greater uncertainty about the design space and less understanding of the processes which could result in product failures that also create risk to the patient due to poor product performance or safety.
The goal of this paper is to continue our illustration of how qualitative and quantitative risk tools can be used in quality-by-design approaches to rationally guide the balance between too many and too few experiments during product development, and to target resources to the factors that can have the greatest impact on patient health (4). This study extends the previous qualitative study and illustrates the use of quantitative Monte Carlo techniques to define the design space and quantitate the uncertainty associated with the design space boundaries. This study will introduce and give practical examples of the Monte Carlo approach in the QbD development process that is outline in ICH Q8, Q9, and Q10.
MATERIALS AND METHODS
Materials
Ciprofloxacin HCl (Lot # 6026) was supplied by R.J. Chemicals, Coral Springs, FL (Manufactured by Quimica Sintetica, Madrid, Spain). Microcrystalline cellulose grades Avicel® PH 102 (Lots # P208819026, P20882001, and P209820744) and Avicel® PH 101 (Lot # P108819435) were donated by FMC Biopolymer (Philadelphia, PA). Pregelatinized corn starch grade Starch 1500® (Lots # IN502268 and IN515968) was obtained from Colorcon (West Point, PA). Hydroxypropyl cellulose (HPC) grades Klucel® EXF (Lots # 99768, 99769, and 89510) was obtained from Aqualon/Hercules (Wilmington, DE). Hydroxypropyl cellulose grade Nisso HPC-L fine (Lot # NHG-5111) was obtained from Nisso America Inc. (New York, NY). Magnesium stearate monohydrate (Lot # MO5676) from vegetable source and magnesium stearate dihydrate (Lot # JO3970) was obtained from Covidien (Hazelwood, MO).
DOE Description
The overall process flow and the primary parameters used for ciprofloxacin manufacturing are given in Fig. 2; these parameters are based upon our previous study (4). For the current study, two separate DOEs were performed to identify the design space: study 1, a small-scale study with a 0.5 kg batch size that examined only processing variables, and study 2, a larger-scale study with a 3.6 or 1.0 kg batch size that examined both processing and formulation variables and their interactions. The base formulation and process conditions, which studies 1 and 2 are built around, are given in Table I, and are based upon our previous study (4). The numerical values for the DOE conditions and results for studies 1 and 2 are given in an Excel® spreadsheet that is provided as supplemental spreadsheet for this manuscript; in this spreadsheet, formulation F5 is the base formulation of Table I.

Overall process design. The mixing, roller compaction, and tableting stages are shown with the process parameters for each stage listed below and the output parameters shown above the stages in italics
For study 1, the process variables study, we used a fractional factorial design that examined three roll pressures (20, 80, and 140 bars), three feed screw speed to roll speed (FSS:RS) ratio(s) of 3:1 (FSS—21 rpm, RS—7 rpm), 5:1 (FSS—35 rpm, RS—7 rpm), and 7:1 (FSS—49 rpm, RS—7 rpm), and the resulting granules were compressed at 8, 12, and 16 kN compression force resulting in a total 15 batches; these are batches F43–F57 in the supplemental spreadsheet.
Study 2 used a fractional factorial design with replicates to examine process variables, formulation variables, and their interactions. Study 2 had two arms; the first arm (study 2a) consisted of 42 batches that were manufactured using an Alexanderwerk® WP120 roller compactor and a Stokes B2 tablet press; the conditions and results of these studies are shown in F1–F42, in the supplemental spreadsheet. The second arm (study 2b) used identical conditions except that a different Alexanderwerk® WP120 roller compactor and a different Stokes B2 tablet press were used at a different location for the granulation and tableting. The tablet tooling was the same in both locations; the test conditions and results of these studies are formulations F58–F68 in the supplemental spreadsheet. For study 2a, the FSS:RS ratio was held constant at 5:1 (FSS—35 rpm, RS—7 rpm), and the roll pressure and the compression force levels were the same as study 1. In addition, the following formulation variables were evaluated: (1) the influence of binder source, i.e., HPC, grade EXF® from Aqualon/Hercules versus Nisso-L HPC manufactured by Nippon Ltd.; (2) lubricant type, magnesium stearate monohydrate versus dihydrate; (3) HPC binder level (2 and 4% w/w); and (4) starch disintegrant level (10 and 14% w/w).
Blending, Roller Compaction, and Tablet Production
Blending
Blending was a two-step process; first, the intragranular components were mixed and then after roller compaction the extra granular components were mixed with the granules. Blending was performed using either an 8 qt or a 16 qt Patterson-Kelly V-blender (East Stroudsburg, PA); both blenders were operated at 30 rpm. For study 1, the 0.5 kg batches (F43–F57) were blended in an 8 qt blender; the intragranular components were blended for 5 min, and the extragranular components were blended for an additional 2 min. For study 2a, the 3.6 kg batches F1–F48 were blended in a 16 qt blender; the intragranular components were blended for 10 min, and the extragranular components were blended for an additional 3 min. For study 2b, the 1.0 kg batches F58–F68 were blended in a 16 qt blender, the intragranular components were blended for 7 min, and extragranular components were blended for an additional 2 min. The intragranular blend contained 54.5% w/w ciprofloxacin and 45.5% w/w excipients (MCC, starch, HPC, and Mg-stearate). The second extragranular blend contained 50% active pharmaceutical ingredient (API) and 50% excipients; half of the starch and Mg-stearate was intragranular and half was extragranular for all formulations.
Roller Compaction
The blends were dry granulated using roller compactor (Model: WP 120 V Pharma, Alexanderwerk Inc., Horsham, PA) equipped with knurled surface rollers; the ribbons were ~25 mm wide. The processing conditions used are described in the Tables I and II. Granulation was performed using a fixed roll-gap of 1.5 mm, and the ribbons were milled in two stages (coarse and fine) using mesh size 10 and 16, respectively. The mill impeller speed was maintained at 50 rpm. The lubricant was combined with a small portion of the other excipients and passed through a 20-mesh wire screen. The roller compactors for studies 2a and 2b were identical models that were operated using the same settings.
Tablets
Tablets were made using a Stokes B2 rotary tablet press fitted with a single set of 11.11 mm (7/16 in) biconcave tooling; the press was operated at 30 rpm for all studies. For all studies, tablets were compressed to a target peak pressure of 8, 12, and 16 kN compression force, see Tables I and II. Tablet weight, thickness, diameter, and crushing strength were consistent in all. The tablet presses for studies 2a and 2b were identical models that were operated using the same settings.
Granule Evaluation
The tapped density (Dt) and bulk density (Db) were measured using JEL Stampf® Volumeter Model STAV 2003 (Ludwigshafen, Germany) and Sargent-Welch (VWR Scientific Products), respectively; methods for both techniques were in accordance with the USP method described in USP <616>. The Carr Index (CI) was calculated as follows:
Granule size was measured by laser diffraction using the Malvern Mastersizer (Malvern Inc., Worcestershire, UK) with a sample size of 5 g operated at an air pressure of 20 psi and a feed rate setting of 2.5. The average mean particle size was D [4,3] and the span, (D90–D10)/D50; the reported values for these parameters are the average of three replicates.
Tablet Evaluation
Dissolution studies were carried out in accordance with the USP monograph for ciprofloxacin HCl, using USP Apparatus II, Model SR8 Plus (Hanson Research; Chatsworth, CA) using 900 mL of 0.01 N HCl at 37 ± 0.5°C and the paddles were operated at 50 rpm. Samples were collected using an autosampler, Autoplus Maximizer (Hanson Research; Chatsworth, CA). The amount of ciprofloxacin HCl released was measured using UV–Visible spectroscopy (Pharmaspec UV-1700, Shimadzu) at 276 nm wavelengths. Disintegration tests were performed on six tablets in accordance with USP method <701> using basket-rack assembly and water as media which was maintained 37 ± 0.5°C. The tablet breaking force was determined using hardness tester (Model HT-300) manufactured by Key International, Inc. (Englishtown, NJ) and the average values of six tablets were reported. Friability tests were performed using Vankel Inc. (Cary, NC) friability apparatus (Model 45-1000) in accordance with USP method <476>. Typically, 6.5 g were analyzed. Content uniformity was performed using weight variation as specified in USP general chapter <905>, and the average value of ten tablets was reported.
Quantitative Methods
To develop regression models for analysis, we used the SAS 9.3 interface, ADX® for Design and Analysis of Experiments (SAS version 9.3, SAS Institute Inc., Cary, NC), as a design-of-experiments (DOE) workbench. For routine statistical analysis and programming, we used SAS, R statistical software, and Microsoft Excel™. Monte Carlo (MC) and Markov chain Monte Carlo (MCMC) simulations were performed using R, Excel, and the Excel add-in, @RISK® (Palisade Corp., Ithaca, NY) for Microsoft Excel. None of the specific software tools were unique for this analysis: the software was chosen for reasons of availability and ease of use (e.g., R and Excel).
The design space can be thought of as a system of multiple regression equations for the dependent variables (y), each as a function of several process variables (X). Each CQA regression can be solved independently; however, the purpose of design space modeling is to find the region for which all of the ys as CQAs meet the target quality profile simultaneously. As a thought experiment, a design problem using process parameters, A, B, and C, might be shown to be significant predictors of the CQAs (y i ) in the system,
where e i are the random errors. The system shows that the factors A, B, C and the interaction, AB, are not predictors occurring evenly in all three equations. This “design space” model might be fit equation-by-equation using various regression methods including ordinary least squares (OLS) after which overlapping ranges of “acceptable” CQA values (y i ) might be inferred graphically or by independent calculations (e.g., ICH Q8(R)). However, there are computational approaches to finding a jointly acceptable design space solution among multiple predictive equations. One such approach is to use MCMC simulation to sample the posterior multivariate distribution of CQAs.
According to Peterson (9), optimizing the process parameters for a design space might be thought of as a statistical reliability problem in which a set of acceptable process parameters, such as those discussed in the authors previous study (4), is developed from conditions in which the probability that the CQA responses (Y = [Y 1 , Y 2 , Y 3 ,…, Y n ]T) are within an acceptable design space (A) exceeds a predetermined reliability, R,
In this equation, x is the vector of process parameter inputs, [x 1 , x 2 , x 3 ,…, x n ]T. The marginal acceptance probability for a CQA, Pr(Y i ∈ A i ), the probability Y i or the estimate of the ith CQA is acceptable, is calculated from the ratio of the number of simulated values of Y i falling within the target design space specifications (A i ), divided by the total number of iterations.
The theory for ordinary least squares (OLS) regressions for a system of equations include an assumptions that the residual errors (e = y − X β) from one CQA to the next are not correlated. In a multivariate design problem, correlations among the CQAs might be expected to occur as the design space is more finely defined. In such cases, one quantitative solution for the system of linear regressions in the presence of cross-equation correlations is “seemingly unrelated regression”; this method is described in (9,20,21). We used either SAS (Proc Syslin) or R package “systemfit” for finding estimates of the regression parameters and the cross-equation covariances and correlations. Once having the prior estimates, MCMC on the SUR model was performed in either R using package “bayesm” or in Excel using our own VBA program. Prior to sampling the poster distributions using MCMC, estimates of the regression parameters and cross-model covariance are needed.
The overall strategy for the quantitative analysis is depicted in Fig. 3 for an arbitrary example of three process parameter inputs and three CQAs. Essentially, the outputs from the multiple regression program provide estimates of the SUR model regression coefficients, standard errors, and the cross-model covariance (Σ); these parameters are subsequently use in the MCMC sampling of the design space. We explored various methods of sampling from the posterior, multivariate region for the CQAs falling within the acceptable “design space.” For example, we implemented an approach similar to Peterson’s for sampling a multivariate normal distribution N(0, I) and multiplying by the Σ 1/2 (or “Cholesky”) square root matrix from the cross-model covariance matrix (9). We used the R bayesm algorithm, “rsurGibbs,” to sample MCMC chains first for β, given Σ, after which updated β are used to sample for new values of Σ, or (Σ |β). The marginal and joint probabilities for the ŷ i or CQAs were calculated according to Pr (ŷ i ∈ A)—the probability that the CQA is within the acceptable region (22). Although convergence of the MCMC sampling was generally possible in ~100 iterations, 250 to 500 iterations were generally used. Once having found the β, a set of x constraints can be calculated according to Eq. 3 above. A more detailed pseudo-algorithm is included in Appendix A.

Experiment modeling flowchart. The regression coefficient and standard errors were obtained from the experimental data and analyses using SAS with the ADX interface. The resulting coefficients and standard errors data were used as regression coefficients and uncertainty in @RISK® to perform Monte Carlo simulations of the output distributions. If the posterior reliability improved, the input (process parameter) distributions were adjusted accordingly. Typically, N = 100 simulations of 600 iterations each were used to find the maximum reliability
Prediction of Optimal Design Space Process Parameters
There are different approaches for initializing the process parameters as simulation inputs. First, a grid of process parameters values can be defined for the purpose of covering the design space and presentation in (e.g.) lattice plots (9). Second, values of process parameters can be drawn from appropriate distributions for each process parameter and CQA equation (Y i ). Finally, process parameters shared across the CQA equations can be sampled from a single set of distributions. All three of the approaches were explored during this work.
For simplicity, a set of simulations began with identical input process parameter distributions: either uniform, normal, or beta-general. The starting lower (θ 1 ) and upper (θ 2 ) distribution limits for either uniform or beta distributions were taken from the minimum and maximum values of the experimental settings. In the case of normal distributions, the parameters of the mean (θ 1 ) and standard error (θ 2 ) were derived by assuming that the experimental minimum and maximum values represented the 5th and 95th percentile values of the normal distribution, respectively. Once parameterized, the iterative Gibbs sampling approach calls for first sampling for β given the cross-model covariance, Σ. After sampling values of (β | Σ) in Monte Carlo chains, the updated β are used to sample for new values of Σ, or (Σ |β); after which, the sampling cycle repeats. If the system of equations is stable, the Monte Carlo chains converge to averages β and Σ estimates.
RESULTS AND DISCUSSION
This study builds upon our previous studies that used risk analysis methods to identify the factors that have the greatest risk of affecting product quality (4). In this earlier study, first, a “cause and effect” or “Ishikawa fishbone” diagram and Failure Mode and Effect Analysis (FMEA) were used to qualitatively identify the most likely material and process variables that could affect the QTPP for the granulation and tablet (23). This was followed by a screening DOE based upon the Plackett-Burman design to quantitatively assess the significance of the variables that were qualitatively identified (24), i.e., to determine if the variables identified by FMEA were really significant. For the current study, we used the parameters identified in our previously study (4) to a put together a DOE from which a response surface model can be built and the design space can be determined to meet expected target conditions and a preset reliability criteria.
The input variables described in this section were chosen based upon the risk analysis carried out in our previous study (4). These input variables are a combination of formulation and process variables. For the formulation variables, we have identified the binder level and source, disintegrant level, and Mg stearate type as the highest risk variables that should be examined when developing the design space. For this formulation, the binder was HPC; our previous results (4) and the literature have shown that physical-chemical properties of HPC (25) and the level in a roller compaction granulation formulation (26) can affect the mechanical properties of a tablet and the drug release rate. Based upon this information, we studied the source and level of HPC; because different sources of HPC are made from different feed stocks, different manufacturing methods and different processing conditions can affect the physical-chemical properties of HPC and hence product quality. Starch was used as a disintegrant for these studies; the starch was always added 50% intra and 50% extra granular with the total level being varied; based upon our previous studies, we found that the level of starch can be important for product quality (4). Mg stearate, one of the highest risk excipients, was used as a lubricant. It is know that properties such as crystal structure of Mg stearate can affect lubricity and the drug release rate from a tablet (27–30), because the Mg Stearate can coat the particles literature during blending (31–33), which reduces tablet tensile strength (31,34–36) and prolongs tablet disintegration and dissolution (33). In addition, it has been shown that the properties of Mg stearate and most other excipients can be variable from lot to lot and from manufacturer to manufacturer (37); this variability could explain differences in the results seen from different studies.
The process variables studied were roll pressure, FSS/RS ratio, and Pmax. For roller compaction, ribbon quality is the key to making good granules, and the three main variables that influence powder consolidation into a ribbon are the rate of powder feed into the roller compression zone, the roller speed which determines how fast powder is removed from the compression zone, and the roll pressure, which controls how much the powder in the compression zone is compressed (38). For tablet compression, the turret speed, roller geometry, and the degree of powder compression in the die are critical to the formation of the tablet properties; to save resources, we have chosen to fix all these variables except Pmax.
Blend uniformity is a critical parameter that affects tablet content uniformity. However, we will not include mixing parameters in the design space because for high-dose drugs like ciprofloxacin (50% w/w in this study), generally, blending is considered a low to moderate risk processing step, and we have implemented a near infrared (NIR) monitoring system to ensure blend homogeneity. The development and application of this system are described in Kona et al. (39).
Granule Properties
A summary of the granule and tablet characteristics are presented in the supplemental spreadsheet. As described previously, batches F1–F42 were manufactured at site 1, which evaluated roll pressures, compression force, and formulation variables such as binder and lubricant type and source on the critical quality attributes of granules and final dosage form. Batches F43–F57 evaluated the influence of roller compaction process parameters such as roll pressure and feed screw speed to roller speed ratio on granule and tablet attributes which was also manufactured at site 1, and batches F58–68 manufactured at site 2.
In general, an increase in roller pressures from 20 to 140 bars increased the average granule size; this was accompanied by a decrease in relative span (spread of granule size distribution). It is well known that increasing in roll pressure produces ribbons with higher tensile strength due to higher degree of material consolidation in the nip region; when these ribbons were milled, the granule size was larger compare to ribbons produced at a lower roll pressure (40). Also, the granule size increased when MgSt-M was replaced with MgSt-D. This behavior could possibly be explained by differences in the particle size and surface area of monohydrate (10.6 μm) and dihydrate forms (14.3 μm). See discussion below for statistical analysis
Examining the data from both manufacturing sites indicates that the granules size obtained at two manufacturing sites are different; this occurred despite efforts to keep the experimental conditions the same at both sites. Given the fact that the formulations and materials used at both sites were the same, a possible reason for this difference could be due to the fact that even though all the settings were identical, there could be calibration differences; thus, the actual parameters could be different. In addition, as mentioned above, Mg stearate and other excipients can be variable, and this variability can cause excipients to behave differently in different situations, so this could also be a contributing factor to these results. Roller compaction process parameter such as feed screw speed to roller speed ratio (3–7) did not influence the particle size under the range tested and was considered insignificant; particle size data is given in the supplemental material associated with this paper.
Tablet Properties
The data indicates that increasing the roll pressure at a given compression force decreases the crushing force of the tablets. This can be explained by loss of compactability or work-hardening phenomenon commonly observed with plastic materials such as microcrystalline cellulose. Several authors have reported that this work-hardening phenomenon results in a pronounced decreased in tensile strength (2,3,23,25,26). In addition, for a given roll pressure, an increase in compression force increased the crushing force of the tablets. It was also observed that increase in the HPC binder level from 2 to 4% significantly increase the crushing force of the tablets. For ciprofloxacin release, roll pressure, compression force, binder levels, and disintegrant levels were found to influence the disintegration time.
The granules manufactured at site 2 were compressed into tablets using an identical rotary press under the same operating conditions. Crushing force and disintegration data were found to be statistically different from the tablets obtained from site 1. The reason for this behavior was described earlier. Similar to granules results, feed screw speed to roller speed ratio was found to be insignificant for crushing force and disintegration time within the range tested; see discussion below for statistical analysis.
Estimation of Process Parameters from Acceptable Design Space Runs
The primary goal of the granulation analysis was to find optimal operating parameters and material inputs. First, the data from the granulation stage were fit to the regression models using SAS as described above. For this analysis, RP, FS:RS, HPC type, MgSt type, HPC type, and Starch 1500 level were regressed against granule size, granule span, bulk density, tapped density, and CI; the results are shown in Fig. 4. The “prediction profiler” plots in Fig. 4 are used in an exploratory data analysis to identify the significant regressions in the design of experiments. In Figs. 4 and 5, the investigator can quickly identify significant regressors individually against each of the proposed outputs or CQAs. Additionally, the plotted confidence intervals (gray-shaded regions) and the regression prediction limits provide visual notion of the uncertainty in each input process attribute and quality attribute output.

Prediction profile for the granulation stage. Representative of the outputs and 95% prediction intervals are shown for bulk density (DB), tapped density (DT), the Carr index (CI), the average particle size (X_AVE), SpanX, and the Hausner ratio, as functions of the process variables, roller pressure (RP), the feed screw-roller speed ratio, HPC source, Mg stearate type, percent EXF, and the 1500 level. The gray-shaded bands are the 95% confidence bands on the output variables and the red lines represent the 95% prediction intervals on the specific regression

Prediction profile for the tableting stage. Representative results are shown. The outputs and 95% prediction intervals are shown for assay weight (WT_AVE), breaking force (BF), dissolution time (DT_AVE), friability (FRIABILI), and the percent dissolved after 45 min (Q45). As functions of the process variables, roller pressure (RP), the feed screw-roller speed ratio, the maximum pressure (Pmax), HPC source, Mg stearate type, percent EXF, and the 1500 level
Table II and Fig. 4 show the results of optimization studies for the granulation stage. These results were used to confirm the previous results before proceeding to the tableting stage. The prediction profiler results confirm both visually and quantitatively that most of the variability observed in the measured properties of this stage was due to roller pressure. The results confirm the qualitative risk assessment performed previously on these components and are shown here as the preliminary staging for simulation of the tableting stage (4). No further simulations or analysis of the granulation stage data were necessary for the tableting stage simulations.
The results of SAS/ADX optimization studies using the tableting data are given in Table III and Fig. 5. Ultimately, roller pressure (RP), maximum compression pressure (Pmax), and the binder source (hydroxypropyl cellulose) grade EXF content most strongly impact the quality attributes of assay weight, breaking force, friability, and disintegration time. Mg stearate, HPC, and starch 1500 level have relatively low impact on the output parameters. Nevertheless, the interactions of these factors revealed by the SAS/ADX exploratory analysis suggested that it is useful to retain the factors in predictive calculations.
The SAS/ADX regression parameters yielding these prediction limits were used in the @RISK® Excel MCMC simulations. Initial results showed that the minimum design space acceptance criterion, R, could be raised from 0.8 to 0.9 without a loss of model performance. Initial results showed that different assumptions in the prior distributions for the process parameters lead to different final reliability; however, all of the assumptions examined lead to model convergence. An example of convergence for beta-general prior distributions is shown in Fig. 6. The typical results in terms of the reliability measure are shown in Fig. 7 for 100 simulations. Typical marginal frequency distributions for the jointly acceptable CQAs are depicted in Fig. 8.

Results of 100 simulations of 600 iterations each on the posterior probability estimate, R. Beta-general prior distributions of the process parameters were assumed at the outset. The starting minimum and maximum values of the beta-general distributions were those of the experimental parameters. In cases in which the solution was slow to converge, the starting parameters were manually adjusted analogously to the “burn-in” process in Gibbs sampling

Results from 100 simulations of the reliability calculation. Joint posterior probability that the tablet weight, breaking force, friability, and disintegration time are within acceptable limits was calculated for each of the 100 simulations of 600 iterations, each

Individual output distributions for the critical quality attributes (CQAs). Once having the optimal process parameters for the simulations, a single simulation of 5000 iterations was run in order to produce the distributions for breaking force, disintegration time, tablet weight, and friability. Numbers above each plot are the 5th and 95th percentile values of the distributions
The simulations show that optimized process parameters could be identified that will exceed a reliability criterion, R ≥ 0.9. The final optimized ranges of process parameters that yield the results are given in Table IV for both the laboratory set and the extended (laboratory + contract manufacturing) set of formulations using beta-general prior distributions. Based on the maximum joint posterior probability for breaking force, friability, and disintegration time, beta prior distributions of process parameters outperformed equivalent MCMC runs that assumed either uniform or normal distributions. In the former instance, the MCMC convergence was extremely slow and the final estimate of R depended on the width of the starting uniform distributions. In the case of normally distributed priors, the 5th percentile estimates were typically well below the starting experimental minimum settings. Although numerically solved, the fact that the lower range extends beyond the experiment suggests that empirical validation would be warranted before adopting the MCMC-derived limits.
Although a robust solution for the design space could be found without sampling the cross-model terms, the complete Gibbs MCMC sampling of SUR covariance in the full model failed in repeated attempts to yield suitable reliabilities for a final design space. The failure to converge at a suitable reliability was likely due to very low cross-model covariance for the system of linear CQA equations. If the off-diagonal covariance terms → 0, the seemingly unrelated regression does not differ from truly unrelated regressions. Our independently sampled CQA regression equations provide a more efficient path to an optimal design space than the slow convergence using the sampling for SUR. Essentially, the simple “overlapping” approach to a design space shown in ICH Q8(R), example 2 of the Annex was adequate for finding a design space (1). Although cross-model Gibbs sampling was not a significant improvement in finding an optimal design space, our use of MCMC to the design space problem ultimately provided estimates of design space uncertainties that are useful for quality risk management.
Finally, one of the target CQAs is dissolution of 80% release in 30 min. The mean ± SD for the extended set of formulations is 86 ± 15 (%) with a median estimate of 91%. The Q30 estimates were not strongly predicted by the independent process parameters; thus, Q30 was dropped in this analysis of the use of independent predictors and MCMC. The use of higher-level interaction terms as predictors, including the dissolution percentages, is a subject of current further study.
Nature of the Results in the Quality Risk Management Paradigm
Reliability in process engineering terms is an incident or time-dependent probability that the unit process is not in a state of failure (41). The probability of an adverse event, defined as “falling outside of the design space,” is logically, (1- R), for a given period of time. Thus, the probability of not meeting the operational design space criteria—a possible risk endpoint—might be estimated from the lower tail of the distribution in Fig. 7 below 0.8. The results suggest that there is a vanishingly small chance of failing below R = 0.8 given the optimal process parameters and conditions for this study. However, this satisfying result comes with the caveat that in a complex multivariate problem solved using Gibbs sampling, there are possibly multiple solutions. Although the repetition of the simulations to generate Fig. 7 addresses overall uncertainty from the propagation of uncertainties from the numerous parameters in the model, this method cannot address model uncertainty that arises from variable selection and structural form of the model. Another caveat with this approach is that the results depend upon the parameter variability used to construct the model from which the Monte Carlo simulations were made. For example, in these studies, we only used one batch of API when developing the regression model; however, if we were to use multiple batches of API, this would inevitably add more variability into the regression model, and this variability could change the Monte Carlo simulation results, which depending upon how sensitive the model was to this parameter could affect the design space boundaries. As with all statistical methods, they are only as reliable as the data used to create the model is representative of the real-world situation to which the model will be applied.
Process reliability, however, is only one part of a “risk equation” for quality risk management. According to ICH Q9, risk is a combination of the probability of adverse events and the severity of the outcomes (2).Footnote 1 Although the design space parameters, controlled largely by three process parameters and monitored using three quality attributes as outputs, describe a very robust process, the severity of a harm that can be linked to a specific quality attribute is needed to fully link this work to patient risk (2). For example, the disintegration time, if failing to meet the design space criteria, could be linked with causing sub-therapeutic doses of the product. Other operational design space failures, such as failed assay weight, might be linked with adverse outcomes from either sub- or super-potent product.
Finally, the present study models the development design space with respect to formulation and developmental process parameters. Taking these posterior estimates for the process parameters into a manufacturing process would logically include physical equipment reliabilities as a dimension of reliability (41). For example, the reliability of the mechanical units (mixers, rollers, tablet press, etc.) over time is part of the overall risk analysis for risk management decisions in the production environment (42).
CONCLUSIONS
The use of statistical methods like Monte Carlo simulation can allow us to associate a risk with the design space boundaries. These boundaries are not absolute in nature, but an expression of relative probability. This approach is a natural follow up to the qualitative methods discussed in the first paper.
Notes
ICH Q9 defines risk as combination of the probability of harms and the severity of the harms (e.g., consequences.) This is a highly generalized definition: more quantitatively-based definitions are necessary for specific design space analyses.
References
ICH. Q8(R2) Pharmaceutical development. Part I: pharmaceutical development, and. Part II: annex to pharmaceutical development. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf. 2009.
ICH. Q9 quality risk management. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf. 2005.
ICH. Q10 pharmaceutical quality system. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Step4/Q10_Guideline.pdf. 2008.
Fahmy R, Kona R, Dandu R, Xie W, Claycamp G, Hoag S. Quality by design I: application of failure mode effect analysis (FMEA) and Plackett–Burman design of experiments in the identification of “Main Factors” in the formulation and process design space for roller-compacted ciprofloxacin hydrochloride immediate-release tablets. AAPS PharmSciTech. 2012;13(4):1243–54.
Claycamp HG. Perspective on quality risk management of pharmaceutical quality. Drug Inf J. 2007;41(3):353–67.
Ahmed R, Baseman H, Ferreira J, Genova T, Harclerode W, Hartman J, et al. PDA survey of quality risk management practices in the pharmaceutical, devices, & biotechnology industries. PDA J Pharm Sci Technol. 2008;62(1):1–21.
Altan S, Bergum J, Pfahler L, Senderak E, Sethuraman S, Vukovinsky KE. Statistical considerations in design space development (Part I of III). Pharm Technol. 2010;34(7):66–70.
Banerjee A. Designing in quality: approaches to defining the design space for a monoclonal antibody process. Bio Pharm Int. 2010;23(6).
Peterson JJ. A Bayesian approach to the ICH Q8 definition of design space. J Biopharm Stat. 2008;18:959–75.
Box GEP, Hunter JS. A confidence region for the solution of a set of simultaneous equations with an application to experimental design. Biometrika. 1954;41:190–9.
Box G, Behnken D. Some new three level designs for the study of quantitative variables. Technometrics. 1960;2(4):455–75.
Box G, Wilson K. On the experimental attainment of optimum conditions. J Roy Stat Soc Ser B (Methodol). 1951;13(1):1–45.
Jain SP, Singh PP, Javeer S, Amin PD. Use of Placket-Burman statistical design to study effect of formulation variables on the release of drug from hot melt sustained release extrudates. AAPS PharmSciTech. 2010;11(2):936–44.
Palamakula A, Nutan MTH, Khan MA. Response surface methodology for optimization and characterization of limonene-based Coenzyme Q10 self-nanoemulsified capsule dosage form. AAPS PharmSciTech. 2004;5(4).
Rasmussen NC. Reactor safety study. An assessment of accident risks in U. S. commercial nuclear power plants. 1975 WASH-1400-MR (NUREG 75/014).
Hubbard DW. How to measure anything: finding the value of intangibles in business. Hoboken: John Wiley and Sons; 2007.
Savage SL. The flaw of averages. Why we underestimate risk in the face of uncertainty. Hoboken: Wiley; 2009.
Gujral B, Stanfield F, Rufino D, editors. Monte Carlo simulations for risk analysis in pharmaceutical product design. Crystal Ball User Conference; 2007.
Kauffman JF, Geoffroy J-M. Propagation of uncertainty in process model predictions by Monte Carlo simulation. Amer Pharm Rev. 2008(July/Aug);75–9.
Zellner A. An efficient method of estimating seemingly unrelated regressions and tests for aggregation bias. J Am Stat Assoc. 1962;57(298):348–68.
Wikipedia. Seemingly unrelated regressions 2014 [cited 2014 May 13]. Available from: http://en.wikipedia.org/wiki/Seemingly_unrelated_regression.
Casella G, George EI. Explaining the Gibbs Sampler. Am Stat. 1992;46(3):167–74.
Ishikawa K. Introduction to quality control. Productivity Press; 1990.
Plackett R, Burman J. The design of optimum multifactorial experiments. Biometrika. 1946;33:305–25.
Picker-Freyer KM, Duerig T. Physical mechanical and tablet formation properties of hydroxypropylcellulose: In: Pure form and in mixtures. AAPS PharmSciTech. 2007;8(Apr):NIL-NIL.
Skinner GW, Harcum WW, Barnum PE, Guo J-H. The evaluation of fine-particle hydroxypropylcellulose as a roller compaction binder in pharmaceutical applications. Drug Dev Ind Pharm. 1999;25(10):1121–8.
Leinonen UI, Jalonen HU, Vihervaara PA, Laine ES. Physical and lubrication properties of magnesium stearate. J Pharm Sci. 1992;81(Dec):1194–8.
Steffens K, Koglin J. The magnesium stearate problem. Manuf Chem. 1993;64(12):16–7.
Swaminathan V, Kildsig D. Effect of magnesium stearate on the content uniformity of active ingredient in pharmaceutical powder mixtures. AAPS PharmSciTech. 2002;3(3):27–31.
Sharpe S, Celik M, Newman A, Brittain H. Physical characterization of the polymorphic variations of magnesium stearate and magnesium palmitate hydrate species. Struct Chem. 1997;8(1):73–84.
Shah AC, Mlodozeniec AR. Mechanism of surface lubrication: influence of duration of lubricant-excipient mixing on processing characteristics of powders and properties of compressed tablets. J Pharm Sci. 1977;66(10):1377–8.
Miller T, York P. Pharmaceutical tablet lubrication. Int J Pharm. 1988;41(1–2):1–19.
Wang J, Wen H, Desai D. Lubrication in tablet formulations. Eur J Pharm Biopharm. 2010;75(1):1–15.
Bossert J, Stains A. Effect of mixing on the lubrication of crystalline lactose by magnesium stearate. Drug Dev Ind Pharm. 1980;6(6):573–89.
van der Watt JG. The effect of the particle size of microcrystalline cellulose on tablet properties in mixtures with magnesium stearate. Int J Pharm. 1987;36(1):51–4.
Kikuta J-I, Kitamori N. Effect of mixing time on the lubricating properties of magnesium stearate and the final characteristics of the compressed tablets. Drug Dev Ind Pharm. 1994;20(3):343–55.
Wang T, Alston KM, Wassgren CR, Mockus L, Catlin AC, Fernando SR, et al. The creation of an excipient properties database to support quality by design (QbD) formulation development. Am Pharm Rev. 2013;16(4):16–25.
Hilden J, Earle G, Lilly E. Prediction of roller compacted ribbon solid fraction for quality by design development. Powder Technol. 2011;213(1–3):1–13.
Kona R, Fahmy R, Claycamp G, Polli J, Martinez M, Hoag S. Quality-by-design III: application of near-infrared spectroscopy to monitor roller compaction in-process and product quality attributes of immediate release tablets. AAPS PharmSciTech. 2015;16(1):202–16.
Lim H, Dave VS, Kidder L, Neil Lewis E, Fahmy R, Hoag SW. Assessment of the critical factors affecting the porosity of roller compacted ribbons and the feasibility of using NIR chemical imaging to evaluate the porosity distribution. Int J Pharm. 2011;410(1–2):1–8.
Modarres M. What every engineer should know about reliability and risk analysis. NY: Marcel Dekker, Inc; 1993.
Ayyub BM. Risk analysis in engineering and economics. Boca Raton: Chapman & Hall/CRC; 2003.
Acknowledgments
The authors would like to acknowledge the financial assistance from the FDA and CIPET. Drs. Bancha Chuasuwan, Ramesh Dandu, and Walter Xie for their technical assistance with the CIP assay. Thanks to Gary Hollenbeck for his assistance with roller compaction work at UPM Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editors: Robin Bogner, James Drennen, Mansoor Khan, Cynthia Oksanen, and Gintaras Reklaitis.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(XLSX 20.0 kb)
Appendix A
Appendix A
Once having modeled the starting process parameters obtain the process parameters, (RP, FS/RS, Pmax, Starch 1500 Level, EXF level), and prior estimates of β and Σ, the MCMC sampling calls for the following steps:
-
1.
For each regression equation (Y i = X β + e i ), calculate new estimates of X from Σ 1/2 ⊗ X where ⊗ is the Kronecker matrix product operation and Σ 1/2 is the Cholesky square root decomposition of the cross-model covariance matrix.
-
2.
First, draw new β given the current cross-model covariance, Σ, or β |Σ
-
3.
Draw from N(0,1) for each of the SAS-obtained coefficients and calculate the current estimate of the coefficients:
$$ {b}_{ij}={\beta}_i+{\sigma}_i\cdot {\left(N\left(0,1\right)\right)}_{ij} $$ -
4.
Calculate \( {y}_j={\widehat{\beta}}_{0,j}+{b}_{1j}{x}_{1j}+\dots +{b_K}_j{x}_{Kj} \) for each output y j (CQA) in the simulation.
-
5.
Repeat steps 1–3 for n iterations.
-
6.
Tally acceptable iterations: calculate marginal and joint probabilities for each of the outputs,
$$ \Pr \left(\left.\mathrm{Acceptable}\right|x\sim distr\left({\theta}_1^{\prime },{\theta}_2^{\prime}\right)\right)=\frac{\mathrm{Count}\ \mathrm{of}\ \left[{y}_j\in {A}_j\left(\mathrm{lower},\mathrm{upper}\right)\right]}{n} $$ -
7.
If the joint probability of acceptance for the CQAs in simulation S, or Pr(Y 1 ,Y 2 , Y 3 , …,Y J )S exceeds the joint probability for the preceding simulation, Pr(Y 1 , Y 2 , Y 3 , …,Y J )S-1, by a defined amount, then update the process parameters distributions by setting new (θ 1 , θ 2 ) for all input process parameter distributions. After visual inspection of simulations, 6–10% improvements of the probabilities were judged to be useful as a threshold for adjusting of the parameters.
-
8.
Repeat 1–6 for S total simulations or until a stable joint probability is achieved.
For our exploratory replication of Peterson’s methods, step 2 was replaced by sampling from a multivariate normal distribution and scaled by the Cholesky square root matrix of the cross-model covariance. Each iteration creates a vector of normal deviates, one for each CQA equation. The deviates were added to the vector of y ij estimates before acceptance-rejection testing of each y ij using the design space targets.
The @RISK® simulations in steps 1–7 were run using a Visual Basic for Applications (VBA) program in Excel that calls @RISK® data objects and procedures. The program used the simulation objects for calculating probability estimates Pr(Y i ∈ A i ) such that the input distribution parameters could be updated. Figure 9 provides a notional view of the adjustments of θ 1 and θ 2 parameters for the input process parameter distributions. For example, a starting uniform distribution for, e.g., roller pressure (RP), could be focused to a narrower, optimized uniform distribution after a few runs of the simulation. The process essentially mimics a Gibbs-like Markov chain simulation having in which all inputs were adjusted given progress toward an acceptable posterior distribution (22). In instances of slow convergence to a maximum posterior probability, the starting parameters were manually adjusted to simulate a Gibbs “burn-in” period.

Sampling distributions for input process parameters. Notional prior and posterior distributions are shown for beta, normal, and uniform priors. The dotted lines show the original distributions and the solid lines show the posterior distribution of input values given an acceptable joint probability for design space CQAs. The beta and uniform distributions were initiated with the minimum and maximum values used in the laboratory experiments. The normal distribution was based on the assumption that minimum and maximum laboratory settings represented the 5th and 95th percentiles
The entire simulation was repeated in order to create a distribution of the posterior joint probability of satisfying the design space criteria or, the reliability criterion, Pr(Y i ∈ A i ) ≥R. For the purpose of this work, 100 simulations of 600 iterations each sufficed to depict distribution of the posterior R distribution.
Rights and permissions
About this article

Cite this article
Claycamp, H.G., Kona, R., Fahmy, R. et al. Quality-by-Design II: Application of Quantitative Risk Analysis to the Formulation of Ciprofloxacin Tablets. AAPS PharmSciTech 17, 233–244 (2016). https://doi.org/10.1208/s12249-015-0349-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0349-2