
Abstract
The nociceptin opioid receptor (NOP), the fourth member of the opioid receptor family, and its endogenous peptide ligand, nociceptin or orphanin FQ (N/OFQ), play a vital role in several central nervous system pathways regulating pain, reward, feeding, anxiety, motor control and learning/memory. Both selective NOP agonists as well as bifunctional agonists at the NOP and mu opioid receptor (MOP) have potential therapeutic applications in CNS disorders related to these processes. Using Surflex-Dock protocols, we conducted a computational structure-activity study of four scaffold classes of NOP ligands with varying NOP-MOP selectivity. By docking these compounds into the orthosteric binding sites within an active-state NOP homology model, and an active-state MOP crystal structure, the goal of this study was to use a structure-based drug design approach to modulate NOP affinity and NOP vs. MOP selectivity. We first docked four parent compounds (no side chain) to determine their binding interactions within the NOP and MOP binding pockets. Various polar sidechains were added to the heterocyclic A-pharmacophore to modulate NOP ligand affinity. The substitutions mainly contained a 1-2 carbon chain with a polar substituent such as an amine, alcohol, sulfamide, or guanidine. The SAR analysis is focused on the impact of structural changes in the sidechain, such as chain length, hydrogen bonding capability, and basic vs neutral functional groups on binding affinity and selectivity at both NOP and MOP receptors. This study highlights structural modifications that can be leveraged to rationally design both selective NOP and bifunctional NOP-MOP agonists with different ratios of functional efficacy.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The nociceptin receptor (NOP), previously known as the opioid receptor-like 1 receptor (ORL1), is the fourth member of the opioid receptor family, and was discovered nearly 25 years ago (1,2). NOP is a G-protein coupled receptor and shares significant homology with μ (MOP), δ (DOP), and κ (KOP) opioid receptors (3). Nociceptin/orphanin FQ (N/OFQ), a heptadecapeptide (FGGFTGARKSARKLANQ), is the endogenous ligand for the NOP receptor and shares significant similarities with the KOP endogenous peptide dynorphin (4,5). Yet N/OFQ has high selectivity for the NOP receptor and does not bind to the other three opioid receptors. NOP receptors are distributed in the central nervous system and periphery and play a significant role in pathways related to pain, drug reward, anxiety, feeding, PTSD, and more (6,7,8,9,10,11,12,13,14,15,16). Numerous pharmaceutical companies, including Astraea Therapeutics, have invested resources to better understand NOP pharmacology with the ultimate goal of developing an approved NOP-targeted drug (17). NOP ligands have been advanced into clinical development as candidate therapeutics for several therapeutic indications—e.g., a NOP antagonist (LY2940094) has been clinically investigated for major depressive disorder and alcohol use disorders (18), a NOP agonist (SCH486757) has been investigated as an anti-tussive (19), whereas a NOP-MOP bifunctional agonist (cebranopadol) has been tested in clinical trials for post-operative pain and neuropathic pain (20,21).
Although MOP agonists are some of the most effective analgesics, their well-known side effects such as respiratory depression and abuse liability have helped fuel the current opioid crisis around the world. Other detrimental side effects that can occur with traditional MOP opioids include opioid-induced constipation, itching sensation (pruritus), as well as opioid-induced hyperalgesia and allodynia. Therefore, there is an unmet need for novel non-addicting analgesics that are devoid of MOP-associated side effects. Notably, NOP activation not only modulates rewarding effects of MOP agonists by inhibiting dopaminergic transmission (22,23) but also synergistically increases MOP receptor-mediated analgesia in the spinal cord (24,25). Thus, we have hypothesized that compounds with bifunctional NOP-MOP agonist activity can potentially produce analgesic effects without MOP-receptor associated reward.
A few research groups including our own have reported NOP ligands with bifunctional or multifunctional efficacy at other opioid receptors. We reported the design and SAR of nonmorphinan NOP-MOP bifunctional agonists AT-201, AT-212, and AT-121, which were characterized for their analgesic and rewarding properties (26,27,28). Colleagues from Grünenthal have reported the discovery of cebranopadol, a mixed nonmorphinan agonist with full agonist efficacy at NOP, MOP, and the delta opioid receptor (DOP), and partial agonist efficacy at the kappa opioid receptor (KOP) (29,30). Husbands and coworkers have developed multifunctional opioid ligands BU08028 and BU10038 based on the morphinan scaffold that bind to NOP as well as the other three opioid receptors (31,32).
As the majority of MOP ligands do not bind to NOP, one strategy to develop NOP-MOP bifunctional compounds is to modify NOP-selective ligands to increase their MOP binding affinity while maintaining affinity at NOP, as we reported previously for AT-212 and AT-121 (28,33,34,35). Most nonpeptide nonmorphinan-type NOP ligands can be dissected into three distinct pharmacophores. The A-pharmacophore contains a heterocycle with a phenyl ring that is either fused or attached via a single bond. The heterocycle is bound to the 4-position of the piperidine ring (B-pharmacophore) either via a single bond or a spirocyclic carbon. Historically, the piperidine ring is a privileged motif in numerous nonpeptidic opioid drug classes as it makes an ionic interaction with a conserved aspartate residue, Asp3.32 in transmembrane helix 3 (TM3) present in all opioid receptors (e.g., Asp1303.32 and Asp1473.32, in the NOP and MOP receptors respectively. The superscript represent the Ballesteros-Weinstein numbering of the residue) (36). The C-pharmacophore in most NOP ligands is bound to the nitrogen of the piperidine ring and is generally a cycloalkyl group with or without varying amounts of aromatic character.
We have published structure activity relationship (SAR) studies on a series of indolinone-derived NOP ligands highlighting the effect of the piperidine nitrogen substituents (C-pharmacophore) on NOP and MOP binding affinity, NOP vs. MOP selectivity, and intrinsic activity at both the receptors (33,34). More recently, we reported a series of C(3)-substituted indoles that were NOP-selective partial agonists (37). Our medicinal chemistry campaign on NOP ligands, supported by structure-based drug design has yielded several selective and bifunctional NOP agonists (35). We have docked both N/OFQ and a known NOP-selective full agonist Ro-64-6198, containing a 1,3,8-triazaspirodecanone scaffold, within a homology model of the active-state NOP receptor, and showed that ECL2 movement of the NOP receptor is critical to ligand-driven NOP activation by agonists (38). The structure-based SAR analysis also explained the increased MOP affinity of 3-alkyl-substituted indolinone NOP ligands (35) and the transformation of a NOP agonist (AT-200) to a NOP antagonist (AT-206) by a single methylene addition in the piperidine nitrogen substituent (39).
Herein, we present a structure-based exploration of four different scaffold classes of NOP ligands and their SAR for NOP affinity, NOP vs. MOP selectivity and intrinsic activity at NOP and MOP receptors. The four classes of NOP ligands contain four distinct heteroaromatic scaffolds in the A-pharmacophore, while having the piperidine ring (B-pharmacophore) and N-4-isopropylcyclohexyl ring (C-pharmacophore) as common pharmacophores. The four scaffolds are of the following chemical classes: (1) indolinones, (2) β-tetrahydrospiroisoquinolinones, (3) 1,3,8-triazaspirodecanones, and (4) indoles. Using an active-state NOP homology model that we developed, as well as an active-state crystal structure of the MOP receptor, we investigated the SAR of a series of side-chain substitutions on the heterocyclic A-pharmacophore of each scaffold to modulate NOP binding affinity, NOP vs. MOP selectivity, and NOP intrinsic activity and provide greater insight and knowledge for future NOP lead candidate design.
MATERIALS AND METHODS
Molecular Docking of Compounds in the Active-State NOP Receptor Model
We previously reported the first active state of the NOP receptor using the only available active-state structure for the GPCR superfamily at the time, the opsin receptor (PDB code: 3CAP) (38). The homology models were built using the “Advance Protein Modeling” module in SybylX 1.2. As the extracellular loop-2 (ECL2) is an integral part of the binding site, the disulfide bridge between TM3 and the second extracellular loop (EL2) was included in the homology model. The details of the model building, loop building, and refinement can be found in our previously published report on homology modeling and molecular dynamics simulation of the NOP receptor (38). A stepwise minimization of the crude model was carried out and was validated using PROCHECK and the ProSA Web Server. This model was utilized in this study to conduct structure-based SAR analysis of the NOP ligands.
All docking experiments were conducted with the active-state NOP receptor model. Compounds were docked into the orthosteric site of NOP using Surflex-Dock. Surflex-Dock is based on the Hammerhead fragmentation/reconstruction algorithm to dock compounds into a defined site. The Surflex-Dock protomol is a precomputed molecular representation of an idealized ligand and represents a negative image of the binding site to which putative ligands are aligned. The structure template used for building the active-state NOP homology model did not contain a ligand. Usually, in such a case, it becomes necessary to use available algorithms for finding putative binding pockets. Instead of using such standard site-finding algorithms, we preferred to use the existing knowledge of the NOP binding site from literature mutagenesis studies (40,41) to locate the orthosteric binding site. Since its discovery, a number of mutagenesis studies on the NOP receptor have identified cognate differences between NOP and the other opioid receptors, as well as residues important for binding with N/OFQ. These studies over the years have identified amino acids such as Asp1303.32, Thr3057.38, and Val2796.51 to be important for binding of NOP. Hence, for this study, the protomol was constructed using a set of active site residues consisting of Tyr581.39, Asp1303.32, Met1343.36, Val2796.51, Thr3057.38, and Tyr3097.42. To optimize the results further, during the Surflex-Dock docking studies, hydrogen atoms (attached to hydroxyl and thiol) and heteroatoms, whose van der Waals surface distances from the docked ligands were < 4 Å in the NOP receptor, were allowed to move to adopt energy-minimized active site conformations of the docked ligands. In addition, the maximum number of starting conformations were kept at four and ring flexibility was also permitted. A maximum of twenty binding poses of each compound were generated. The docked poses were ranked according to the “Total Score” in Sybyl’s Surflex-Dock docking suite and the binding pose with the best score (Table S1) was selected for each compound to compare the binding interactions below.
Molecular Docking of Compounds in the Active State MOP Crystal Structure (5C1M)
Compounds were docked into the orthosteric site of MOP using Surflex-Dock. The protomol for defining the binding site for the docking studies was generated using co-crystallized morphinan ligand BU72. Similar to the molecular docking in the NOP receptor, further optimization occurred during the Surflex-Dock docking studies in which hydrogen atoms (attached to hydroxyl and thiol) and heteroatoms whose van der Waals surface distances from the docked ligands < 4 Å in the MOP receptor, were allowed to move to adopt energy-minimized conformations of the docked ligands at the active site. In addition, the maximum number of starting conformations for each ligand were kept at four and ring flexibility was also permitted. A maximum of twenty binding poses of each compound were generated and evaluated for possible interactions with binding site. The docked poses were ranked according to the “Total Score” in Sybyl’s Surflex-Dock docking suite and the binding pose with the best docking score (Table S1) was selected for each compound to compare the binding interactions below.
RESULTS AND DISCUSSION
To explore the pharmacophores necessary for binding affinity at NOP and MOP receptors and MOP/NOP selectivity, we docked four classes of agonists into the NOP active-state homology model and MOP-active state crystal structure. The basic piperidine nitrogen (B-pharmacophore), common in all compounds studied here, makes ionic interactions with the acidic residues in NOP (Asp1303.32) and MOP (Asp1473.32) receptor binding sites (Fig. 1). This is a key anchoring interaction for all opioid ligands, that involves the conserved Asp in TM3 of all four opioid receptors and a corresponding basic nitrogen pharmacophore present in opioid ligands. For all the NOP agonists reported here, the top docking poses in the NOP and MOP receptors orients the lipophilic N-4-isopropylcyclohexyl group (C-pharmacophore) toward the intracellular end of the ligand binding pocket, allowing for hydrophobic interactions with conserved residues such as Met1343.36 and Met1513.36, and Trp2766.48 and Trp2936.48 in NOP and MOP receptors, respectively. The heteroaromatic A-pharmacophore of these analogs are positioned toward the extracellular end of the ligand binding pocket. Additionally, at both NOP and MOP receptors, these heterocyclic moieties interact with polar and nonpolar residues at the extracellular ends of TM1, TM2, TM3, TM7 and in the case of NOP, acidic residues Glu194 and Glu199 in ECL2.

Docking poses of (a) 1,3,8-triazaspirodecanone 3a (silver) and β-tetrahydrospiroisoquinolinone 2a (pink), and (b) indolinone 1a (cyan) and indole 4a (blue) in the ligand binding pocket of the active state NOP receptor model. Docking poses of (c) triazaspirodecanone 3a (silver) and β-tetrahydrospiroisoquinolinone 2a (magenta), and (d) indolinone 1a (cyan) and indole 4a (green) in the ligand binding pocket of the crystal structure of the active state MOP receptor
We initially docked the four unsubstituted parent compounds of each scaffold class in the NOP receptor. Table I shows the structures and binding affinities of the unsubstituted NOP ligands–indolinone 1a, β-tetrahydrospiroisoquinolinone 2a, triazaspirodecanone 3a, and indole 4a. Docking results show that the amide nitrogen of triazaspirodecanone 3a and β-tetrahydrospiroisoquinolinone 2a acts as a hydrogen bond donor and forms a H-bond with Tyr3097.42 of TM7 (Fig. 1a). In contrast, the carbonyl oxygen of indolinone 1a acts as a H-bond acceptor and makes a H-bond with Thr3057.38 of TM7 (Fig. 1b), whereas the indole pharmacophore in compound 4a lacks any H-bonding capability. This possibly accounts for indole 4a having lower binding affinity than indolinone 1a at the NOP receptor than the other three ligands (Table I). When analyzing all four ligands, the key difference between their binding poses is the extent to which the phenyl ring (in A-pharmacophore) extends into the hydrophobic pocket that consists of Val1263.28 (TM3), Ile1273.29 (TM3), Leu1042.57 (TM2), and Cys200 (EL2) (Fig. 1a). The freely rotating phenyl ring in triazaspirodecanone 3a fits deeper into the hydrophobic pocket than the fused phenyl ring of β-tetrahydrospiroisoquinolinone 2a. We hypothesize that this difference is responsible for the ~ 200-fold difference in NOP binding affinity for triazaspirodecanone 3a (NOP Ki = 0.09 nM), as compared to β-tetrahydrospiroisoquinolinone 2a (NOP Ki = 20.2 nM). In addition, it is possible that the inability of compound 2a to fill the hydrophobic pocket does not enable the change in receptor conformation needed for full NOP activation, thus leading to only partial NOP activation, whereas triazaspirodecanone 3a is a potent NOP full agonist (Table I).
If fully occupying the hydrophobic pocket mentioned above and having a H-bond interaction between the heterocyclic A-pharmacophore and the extracellular portion of the orthosteric binding site in NOP allows for high NOP binding affinity, then triazaspirodecanone 3a satisfies both interactions, β-tetrahydrospiroisoquinolinone 2a and indolinone 1a satisfy only the latter interaction, whereas indole 4a satisfies neither. Yet, indole 4a has better NOP binding affinity than β-tetrahydrospiroisoquinolinone 2a (Table I). In addition, although indolinone 1a only satisfies one of the hypothetical requirements from above, it is a NOP full agonist and nearly as potent as triazaspirodecanone 3a (Table I), which satisfies both hypothetical requirements. These differences are likely due to other variables, such as the different H-bond interactions, and the difference in the junction between the heterocyclic A-pharmacophore and the piperidine ring (B-pharmacophore) affecting the flexibility and ability to rotate into a higher binding conformation.
At the MOP receptor orthosteric site, the A-pharmacophore moieties of all four parent compounds do not make any polar interactions (Fig. 1 c and d). The phenyl rings of triazaspirodecanone 3a and indolinone 1a occupy the hydrophobic subpocket comprised of residues Ile1443.29 (TM3), Val1433.28 (TM3), Trp133 (EL1), and Cys2173.55 (EL2). Interestingly, the docked pose of the high affinity MOP ligand fentanyl in the MOP receptor also shows that fentanyl’s aniline ring occupies the same hydrophobic pocket (Figure S1). It has also been previously reported that the phenyl ring in DAMGO’s N-Me-Phe4 residue occupies the same subpocket (42,43). Hence, both triazaspirodecanone 3a (MOP Ki = 6.81 nM) and indolinone 1a (MOP Ki = 8 nM) (Table I) have good binding affinity at the MOP receptor. However, due to stronger binding at the NOP receptor for triazaspirodecanone 3a compared to indolinone 1a, NOP selectivity over MOP varies widely for both the compounds (76-fold vs. 2-fold, respectively). On the other hand, β-tetrahydrospiroisoquinolinone 2a does not extend deep enough into the hydrophobic gap between Ile1443.29 and Val1433.28 to create optimum hydrophobic interactions, resulting in lower binding affinity of this scaffold at the MOP receptor compared to triazaspirodecanone 3a and indolinone 1a. Indole 4a has the lowest MOP binding affinity of all the parent compounds, which could be due to (a) the phenyl ring being in a flipped orientation that positions it away from Ile1443.29 and Val1433.28, and (b) the energetically demanding conformation adopted by the piperidine ring containing 1,4-diaxial substitution (Fig. 1d). The poor MOP binding affinity of indole 4a helps makes it a selective NOP partial agonist (NOP vs. MOP > 30-fold, Table I), whereas close analog indolinone 1a has much higher MOP binding affinity and is a NOP-MOP bifunctional agonist.
An examination of the extracellular ends of TM1 and TM7 for both NOP and MOP receptors reveals several polar amino acids. Therefore, we hypothesized that if we introduced polar substituents on the amide nitrogen of triazaspirodecanone 3a and β-tetrahydrospiroisoquinolinone 2a, or on the C(3)-position of indolinone 1a and indole 4a, we could affect and alter the binding affinity, NOP vs. MOP selectivity, and potency at both receptors. Consequently, we designed, synthesized, and tested a series of analogs on each scaffold to further our structure-based SAR knowledge with the goal of developing next-generation NOP selective and NOP-MOP bifunctional compounds.
Indolinone Series
The indolinone series contains a carbonyl in the A-pharmacophore that we hypothesized would form a H-bond with Thr3057.38 in the NOP receptor. Unsubstituted indolinone 1a is a bifunctional ligand (NOP vs. MOP ~ 2) that binds with comparable affinity at both NOP and MOP receptors and is a full agonist at both receptors with similar potency (Table II). Indolinone 1a has similar MOP binding affinity as triazaspirodecanone 3a, yet its NOP binding affinity is 44-fold less than compound 3a, likely due to the fused phenyl ring (indolinone) not filling the same hydrophobic pocket that triazaspirodecanone 3a is able to fill. Another possible reason is that the spirocyclic junction in the triazaspirodecanone scaffold fits better into the NOP binding pocket when compared to indolinone, possibly due to the indolinone nitrogen being bound to the piperidine ring via a single bond, allowing for more flexibility that, in this case, is detrimental to NOP binding affinity. Substitution on the indolinone ring at C(3) had a variable effect on the SAR for NOP affinity and NOP-MOP selectivity. Carboxamide derivative 1b due to additional hydrogen bonding capability showed better docking scores (NOP = − 12. 35 kcal/mol and MOP = − 8.57 kcal/mol) compared to the parent indolinone 1a (NOP = − 10.79 kcal/mol and MOP = − 7.74 kcal/mol). It also translated into improved binding affinity for 1b at both NOP and MOP, while retaining the moderate NOP-MOP selectivity of the parent indolinone 1a. Indolinone 1c, with the N-acetyl ethyleneamino side-chain showed greater improvement in the NOP-MOP selectivity, while indolinone alcohol 1d slightly favors binding at MOP over NOP.
The docked poses of C(3)-hydroxyethyl indolinone 1d and C(3)-(N-acetyl)ethylamino indolinone 1c in the NOP ligand binding site show a different orientation for the indolinone ring compared to parent compound 1a (Fig. 2). Unlike 1a, the carbonyl group in C(3)-hydroxyethyl indolinone 1d forms a H-bond with Tyr581.39 instead of Thr3057.38. The phenyl ring of C(3)-hydroxyethyl indolinone 1d is pushed deeper into the hydrophobic pocket due to the H-bond made by the hydroxyl group with the backbone of Leu1042.57. Thus, the docking score of C(3)-hydroxyethyl indolinone 1d (− 12.67 kcal/mol) is better compared to the parent compound 1a (− 10.79 kcal/mol), resulting in slightly higher NOP binding affinity compared to indolinone 1a. The C(3)-(N-acetyl)ethylamino indolinone1c binds at NOP with the indolinone ring flipped 180° horizontally, resulting in the 3-position substituent extending the methyl group deep into the hydrophobic pocket resulting in slightly better binding affinity compared to indolinone 1a.

Docking poses of (a) indolinone 1d (purple) and indolinone 1a (cyan) and (b) indolinone 1c (gray) and indolinone 1a (cyan) in the ligand binding pocket of the homology model of active state NOP receptor
β-Tetrahydrospiroisoquinolinone Series
The SAR of the A-pharmacophore side-chain substituents for the β-tetrahydrospiroisoquinolinone class of NOP ligands is shown in Table III. To improve binding affinity at both the NOP and MOP receptors, we introduced various polar groups onto the amide nitrogen of the β-tetrahydrospiroisoquinolinone ring, such as basic nitrogens in compounds 2b and 2c, and non-basic polar substituents in compounds 2d and 2e (AT-121) (Table III). These analogs have poor NOP vs. MOP selectivity (< 10-fold), except in the case of N-ethylguanidinyl β-tetrahydrospiroisoquinolinone 2c (37-fold NOP selective). The ethylguanidinyl group in 2c significantly enhances its NOP binding, with NOP affinity for guanidine 2c being > 20-fold higher than that of β-tetrahydrospiroisoquinolinone 2a. The guanidinyl group may make ionic interactions with the acidic residues Glu194 and Glu199 found on ECL2 of the NOP receptor. Interestingly, compound 2c showed the best docking score at NOP receptor, − 14.18 kcal/mol, compared to parent compound 2a (NOP docking score = − 10.53 kcal/mol) and other compounds in the β-tetrahydrospiroisoquinolinone series.
Functionally, the potencies of N-ethylamino β-tetrahydrospiroisoquinolinone 2b and N-ethylguanidinyl β-tetrahydrospiroisoquinolinone 2c are roughly the same as parent 2a, and they are all partial agonists at the NOP receptor. The inability of the β-tetrahydrospiroisoquinolinone scaffold to provide NOP full agonists implies that their conformations within the NOP binding pocket prevents the receptor movement needed for full activation. Although there is a ~ 5–10-fold increase in NOP binding affinity for N-ethylamino 2b and N-ethylguanidinyl 2c, possibly due to the ionic interaction leading to improved NOP binding affinity, non-ionic polar substituents such as N-ethylacetamide β-tetrahydrospiroisoquinolinone 2d has reasonable NOP binding affinity, while N-ethylsulfamide β-tetrahydrospiroisoquinolinone 2e has a stronger NOP binding affinity compared to parent β-tetrahydrospiroisoquinolinone 2a. In addition, the contribution of the ionic interactions of the A-pharmacophore substituents leading to high NOP affinity is further confirmed by the loss of NOP affinity when the amino side chain in β-tetrahydrospiroisoquinolinone 2b (NOP docking score: − 13.60 kcal/mol) is converted to acyl amide in 2d (NOP docking score: − 12.93 kcal/mol). Nevertheless, this 5-fold loss of NOP binding affinity is the smallest compared to the SAR observed in other scaffolds: > 10-fold for triazaspirodecanone (Table IV), and > 20-fold for indole (Table V).
We recently showed in primates that 2e (AT-121) is a strong analgesic that provides antinociceptive effects similar to morphine yet produces no opioid-induced side-effects such as respiratory depression or abuse liability. In addition, it was shown that 2e (AT-121) inhibits oxycodone self-administration, as well as inhibits reinforcing effects in monkeys (28). Our modeling suggests that the sulfamide group in 2e (AT-121) makes a polar H-bond network within the NOP receptor with several polar residues situated on the extracellular end of the ligand binding site for both NOP (Glu194, Glu199, and Thr3057.38) and MOP receptors (Asn1272.63, Gln1242.60, and Tyr751.39) (Fig. 3a and b), leading to high binding affinity at both receptors (Fig. 3). Compound 2e also showed significantly better docking scores at NOP (− 13.66 kcal/mol) and MOP (− 11.22 kcal/mol) compared to the parent compound 2a (NOP docking score: − 10.53 kcal/mol and MOP docking score: − 8.36 kcal/mol) due to better binding interactions with both the receptors.

Docking poses of a β-tetrahydrospiroisoquinolinone 2e (AT-121) (yellow) in the ligand binding pocket of the homology model of active state NOP receptor and b β-tetrahydrospiroisoquinolinone 2e (AT-121) (green) in the ligand binding pocket of the crystal structure of the active state MOP receptor
1,3,8-Triazaspirodecanone Series
The triazaspirodecanone scaffold provides ligands with high-binding affinity, potency, and intrinsic activity for the NOP receptor (Table IV). The parent triazaspirodecanone 3a is a NOP full agonist, MOP partial agonist with the highest NOP vs. MOP selectivity. It was found that substitution of the amide nitrogen in the A-pharmacophore leads to a significant decrease in NOP-MOP selectivity due to a reduction in NOP binding affinity. Substitution with basic nitrogen-containing moieties, such as in 3b and 3c, retains a high NOP binding affinity, resulting in only a small loss in NOP selectivity. Compounds 3b and 3c both showed very high docking scores (− 15.83 kcal/mol and − 16.10 kcal/mol) at NOP receptor compared to other compounds reported in this study, which correlated well with the higher binding affinities observed at the NOP receptor for both the compounds. In addition, both potency and intrinsic activity of 3b and 3c at the NOP receptor is very similar to the unsubstituted triazaspirodecanone 3a, albeit with reduced stimulation of the MOP receptor.
The substituents with basic functional groups on the A-pharmacophore can make ionic interactions with the NOP receptor ECL2 residues Glu194 and Glu199, resulting in high NOP binding affinity. In the case of compound 3c, the guanidinium group in the A-pharmacophore substituent not only makes ionic interactions with acidic residues on ECL2, but also H-bonds with the backbone of Leu301 (Fig. 4a). Additionally, the carbonyl group in 3c makes a H-bond interaction with Thr3057.38 residue, which also contributes to higher NOP binding affinity. In contrast to the stronger ionic interactions of guanidinium group at the NOP receptor, 3c makes only H-bonding interactions with the neutral polar residues such as Asn1272.63 (TM2) and Tyr751.39 (TM1) in the MOP receptor binding site (Figure S2). This allows 3c to maintain good selectivity for NOP over the MOP receptor (NOP vs. MOP selectivity ~ 47-fold). Alternatively, compound 3e, which contains a polar sulfamide group and can act as both hydrogen bond donor and acceptor, provides a bifunctional profile (NOP vs. MOP ~ 1.1-fold).

Docking poses of (a) triazaspirodecanone 3c (magenta) and (b) triazaspirodecanone 3e (yellow) in the ligand binding pocket of the homology model of active state NOP receptor
Our modeling suggests that the sulfamide group in compound 3e makes H-bond interactions with Glu194 on the NOP ECL2, and Asn1272.63 and Tyr751.39 of the MOP receptor (Figure S2), which results in good docking scores (NOP docking score: − 13.52 kcal/mol and MOP docking score: − 11.28 kcal/mol) and binding affinities at both receptors, albeit with reduced binding affinity at NOP compared to guanidine-containing 3c. Similarly, 3d and 3f with neutral polar substituents such as amide and alcohol groups, respectively, have a dual NOP-MOP binding profile resulting from H-bonding interactions with polar residues of both receptors (Table IV).
It is interesting to note that both an alcohol (1d) and acylamide (1c) substituent on the indolinone scaffold slightly improves NOP binding affinity compared to unsubstituted indolinone 1a (Table II), while the same substituents on the triazaspirodecanone scaffold (3d and 3f) lead to > 25-fold loss of NOP binding affinity compared to unsubstituted triazaspirodecanone 3a (Table IV). With respect to the triazaspirodecanone and β-tetrahydrospiroisoquinolinone series (Tables III and IV), the neutral polar substituents on the heterocyclic A-pharmacophore generally lead to NOP-MOP bifunctional profile, whereas the positively charged functional groups generally produce NOP selective ligands. In addition, the larger sized β-tetrahydrospiroisoquinolinones appear to be partial agonists at the NOP receptor in comparison to the triazaspirodecanone class of compounds, which are generally potent full agonists at NOP. This could be due to the inability of the fused phenyl ring in the β-tetrahydrospiroisoquinolinone scaffold to occupy the hydrophophobic pocket residing between TM2 and TM3, compared to the pendant N-phenyl ring of the triazaspirodecanone series.
Indole Series
The indole scaffold (A-pharmacophore) shows more variability in the effects of substituents for both NOP binding affinity and intrinsic activity, when compared to the triazaspirodecanone scaffold. For instance, unsubstituted indole 4a has NOP binding affinity almost 100-fold lower than that of triazaspirodecanone 3a (Table V). Interestingly, the difference in the docking score of triazaspirodecanone 3a (− 11.55 kcal/mol) versus indole 4a (− 12.08 kcal/mol) at NOP receptor did not appear to correlate with the 100-fold difference in binding affinity observed for these two scaffolds. The majority of the highest scoring docking poses of scaffold 4a show that the fused phenyl ring of the indole A-pharmacophore does not occupy the hydrophobic pocket at the extracellular end of the NOP binding pocket, which we hypothesize is important for ligand-induced NOP activation. Furthermore, unsubstituted indole 4a does not make any H-bonding or polar interactions in the NOP binding pocket, leading to lower NOP affinity and only partial agonist activity compared to the spirocyclic 1,3,8-triazaspirodecanone and the indolinone scaffolds, which are generally NOP full agonists. Thus, we focused on adding polar functionalities to the indole ring to improve NOP affinity for this series. Adding a positively charged ionic moiety in the A-pharmacophore on the C(3) position of the indole improved binding affinity at NOP as seen with the C(3)-methyleneamino indole 4b and ethyleneamino indole 4c (NOP Ki = 2 and 3 nM, respectively). Compounds 4b and 4c both also showed better docking scores at NOP receptor (− 12.91 kcal/mol and − 13.30 kcal/mol) compared to the parent compound 4a (NOP docking score: − 12.08 kcal/mol).
The importance of these ionic interactions at NOP for improving binding affinity of the indole and other series is further confirmed by the loss of affinity of the acylated amino analog 4d, and the modest binding affinity of the polar but nonionic C(3)-hydroxymethyl indole analog 4e. Indole 4e however makes a polar H-bond interaction with Tyr 3097.42 (Fig. 5). Notably, the C(3) substituted indole analogs 4b-4e are all partial agonists at NOP. At the MOP receptor, the C(3) substituted indoles 4a-e make weak interactions and show poor MOP binding affinity (Table IV). The C(3) substituted indole series of NOP ligands therefore yield NOP-selective partial agonist ligands.

Docking poses of (a) indole 4e (yellow) and indole 4f (AT-312) (blue) in the ligand binding pocket of the active state NOP receptor model and (b) indole 4e (yellow) and indole (4c) (purple) in the ligand binding pocket of the crystal structure of the active state MOP receptor
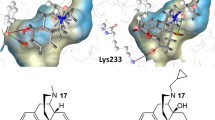
Interestingly, computational modeling predicted that a C(2) substitution on the indole A-pharmacophore may afford a binding conformation that resides deeper into the NOP TM2-TM3 hydrophobic pocket and retains the other key interactions at the NOP binding site. The C(2)-hydroxymethyl indole analog 4f (AT-312) forms a H-bond with Tyr3097.42 and the key interaction with Asp1303.32, and occupies the hydrophobic pocket, lined with Val1263.28, Ile1273.29, Leu1042.57, and Cys200. Therefore, the C(2)-analog 4f showed better docking score (− 12.76 kcal/mol) compared to the C(3)-isomer 4e (− 11.92 kcal/mol) and parent compound 4a (− 12.08 kcal/mol) at the NOP receptor. As predicted, indole 4f (AT-312) is a full NOP agonist at NOP (Table V) with subnanomolar NOP binding affinity and it also has high agonist potency compared to the C(3) indoles in Table V. Interestingly, the binding and functional profile of 4f (AT-312) is remarkably similar to triazaspriodecanone 3a (Table IV). We have shown that NOP full agonist 4f (AT-312) attenuates ethanol, morphine, and cocaine rewarding effects in mice (44,45), and we continue to investigate C(2)-substituted indole-series NOP full agonists for various substance abuse disorders.
In the MOP receptor, docked conformations of C(3)-substituted indole alcohol 4e and C(2)-substituted indole alcohol 4f (AT-312), as well as C(3) indole amines 4b and 4c show a H-bond interaction with EL2 residues Cys2173.55 (backbone) or Asp216 (Fig. 5b and Figure S3). To maintain this interaction with ECL2, the 4-isopropylcyclohexyl group of the indole ligands is pushed deeper into the MOP binding pocket. The 4-isopropylcyclohexyl ring (C-pharmacophore) is surrounded by bulky hydrophobic residues (Met1513.36, Val3006.55, and Ile2966.51), and it appears Met1513.36 (shown as spheres in Fig. 5b) has a steric clash with the 4-isopropylcyclohexyl C-pharmacophore of C(3)-alcohol 4e and amine 4c, possibly forcing the cyclohexane ring to adopt an energetically disfavored twisted boat conformation, whereas higher affinity NOP ligands, amine 4b and C(2)-alcohol 4f (AT-312), appear to dock with a chair conformation.
Molecular docking of these four different chemical series of NOP agonists shows several common NOP binding site interactions that afford high NOP affinity. Comparison of the docking poses and binding interactions among the four scaffolds also explains possible reasons for NOP partial agonist activity of the tetrahydroisoquinolinone and the C(3) substituted indole series, compared to the full agonist activity of the 1,3,8-triazaspirodecanone and indolinone series. Notably, however, all four series of NOP agonists were found to bind in a consistent orientation in the NOP binding pocket, wherein the heterocyclic A-pharmacophore was oriented toward the extracellular end of the NOP binding pocket, and participate in a hydrogen-bonding polar network involved in NOP activation (Figs. 1–6) (see also (38)). In contrast to the docked orientation of NOP agonists reported here, the binding orientation of NOP antagonists found in the reported antagonist-bound crystal structures of the NOP receptor (PDB Code: 4EA3, 5DHH, and 5DHG) (46,47) appear to be flipped in orientation, with the heterocyclic A-pharmacophore oriented toward the lipophilic intracellular end of the ligand binding pocket of the NOP receptor and the C-pharmacophore, on the central N-piperidine moiety, oriented toward the extracellular end. This inverted orientation observed for NOP antagonists C-24, SB-612111, and C-35 crystallized in the NOP receptor, is likely due to two main factors—(1) these NOP antagonists contain no polar substituents on their respective heterocyclic A-pharmacophore that would promote favorable hydrogen-bond (H-bond) interactions at the extracellular end of the NOP binding site, and (2) most NOP antagonists have larger hydrophobic C-moieties that may not fit into the hydrophobic pocket closer to the intracellular end of the binding pocket.
Conclusions
Herein, we report molecular docking and structure-based SAR of the heterocyclic pharmacophores of four NOP ligand scaffolds. We analyzed these four NOP ligand series within the orthosteric sites of the NOP and the MOP receptor with the goal of using structure-based design for obtaining high NOP affinity and modulating NOP vs. MOP selectivity. The binding affinity and functional characterization of the NOP ligands revealed several key trends from computational SAR analysis. (1) Substitution in the heterocyclic A-pharmacophore with a basic functional group such as an amine or guanidine generally increases NOP binding affinity and increases NOP selectivity over MOP due to an ionic interaction between the protonated nitrogen and either Glu194 or Glu199 of the NOP ECL2, and (2) ligand interaction with the hydrophobic pocket that resides between TM2 and TM3 near the extracellular end of the NOP binding site appears to be important for high NOP affinity and full receptor activation.
References
Bunzow JR, Saez C, Mortrud M, Bouvier C, Williams JT, Low M, et al. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a mu, delta or kappa opioid receptor type. FEBS Lett. 1994;347(2-3):284–8.
Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, et al. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994;341(1):33–8.
Fukuda K, Kato S, Mori K, Nishi M, Takeshima H, Iwabe N, et al. cDNA cloning and regional distribution of a novel member of the opioid receptor family. FEBS Lett. 1994;343(1):42–6.
Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377(6549):532–5.
Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, et al. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270(5237):792–4.
Chen Y, Fan Y, Liu J, Mestek A, Tian M, Kozak CA, et al. Molecular cloning, tissue distribution and chromosomal localization of a novel member of the opioid receptor gene family. FEBS Lett. 1994;347(2-3):279–83.
Narita M, Mizoguchi H, Oji DE, Dun NJ, Hwang BH, Nagase H, et al. Identification of the G-protein-coupled ORL1 receptor in the mouse spinal cord by [35S]-GTPgammaS binding and immunohistochemistry. Br J Pharmacol. 1999;128(6):1300–6.
Sandin J, Georgieva J, Schott PA, Ogren SO, Terenius L. Nociceptin/orphanin FQ microinjected into hippocampus impairs spatial learning in rats. Eur J Neurosci. 1997;9(1):194–7.
Andero R. Nociceptin and the nociceptin receptor in learning and memory. Prog Neuro-Psychopharmacol Biol Psychiatry. 2015;62:45–50.
Jenck F, Moreau JL, Martin JR, Kilpatrick GJ, Reinscheid RK, Monsma FJ Jr, et al. Orphanin FQ acts as an anxiolytic to attenuate behavioral responses to stress. Proc Natl Acad Sci U S A. 1997;94(26):14854–8.
Gavioli EC, de Medeiros IU, Monteiro MC, Calo G, Romao PR. Nociceptin/orphanin FQ-NOP receptor system in inflammatory and immune-mediated diseases. Vitam Horm. 2015;97:241–66.
Kiguchi N, Ding H, Kishioka S, Ko MC. Nociceptin/Orphanin FQ Peptide Receptor-Related Ligands as Novel Analgesics. Curr Top Med Chem. 2020;20(31):2878–88.
Mercatelli D, Pisano CA, Novello S, Morari M. NOP receptor ligands and Parkinson's disease. Handb Exp Pharmacol. 2019;254:213–32.
Ciccocioppo R, Borruto AM, Domi A, Teshima K, Cannella N, Weiss F. NOP-Related Mechanisms in Substance Use Disorders. Handb Exp Pharmacol. 2019;254:187–212.
Mouledous L. The nociceptin/orphanin FQ system and the regulation of memory. Handb Exp Pharmacol. 2019;254:259–78.
Kiguchi N, Ding H, Ko MC. Therapeutic potentials of NOP and MOP receptor coactivation for the treatment of pain and opioid abuse. J Neurosci Res. 2020.
Zaveri NT. Nociceptin Opioid Receptor (NOP) as a Therapeutic Target: Progress in Translation from Preclinical Research to Clinical Utility. J Med Chem. 2016;59(15):7011–28.
Witkin JM, Wallace TL, Martin WJ. Therapeutic approaches for NOP receptor antagonists in neurobehavioral disorders: clinical studies in major depressive disorder and alcohol use disorder with BTRX-246040 (LY2940094). Handb Exp Pharmacol. 2019.
Woodcock A, McLeod RL, Sadeh J, Smith JA. The efficacy of a NOP1 agonist (SCH486757) in subacute cough. Lung. 2010;188(Suppl 1):S47–52.
Eerdekens MH, Kapanadze S, Koch ED, Kralidis G, Volkers G, Ahmedzai SH, et al. Cancer-related chronic pain: investigation of the novel analgesic drug candidate cebranopadol in a randomized, double-blind, noninferiority trial. European journal of pain (London, England). 2018.
Scholz A, Bothmer J, Kok M, Hoschen K, Daniels S. Cebranopadol: a novel, first-in-class, strong analgesic: results from a randomized phase IIA clinical trial in postoperative acute Pain. Pain Phys. 2018;21(3):E193–e206.
Di Giannuario A, Pieretti S, Catalani A, Loizzo A. Orphanin FQ reduces morphine-induced dopamine release in the nucleus accumbens: a microdialysis study in rats. Neurosci Lett. 1999;272(3):183–6.
Murphy NP, Ly HT, Maidment NT. Intracerebroventricular orphanin FQ/nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetized rats. Neuroscience. 1996;75(1):1–4.
Cremeans CM, Gruley E, Kyle DJ, Ko MC. Roles of mu-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J Pharmacol Exp Ther. 2012;343(1):72–81.
Hu E, Calo G, Guerrini R, Ko MC. Long-lasting antinociceptive spinal effects in primates of the novel nociceptin/orphanin FQ receptor agonist UFP-112. Pain. 2010;148(1):107–13.
Khroyan TV, Zaveri NT, Polgar WE, Orduna J, Olsen C, Jiang F, et al. SR 16435 [1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one], a novel mixed nociceptin/orphanin FQ/mu-opioid receptor partial agonist: analgesic and rewarding properties in mice. J Pharmacol Exp Ther. 2007;320(2):934–43.
Toll L, Khroyan TV, Polgar WE, Jiang F, Olsen C, Zaveri NT. Comparison of the antinociceptive and antirewarding profiles of novel bifunctional nociceptin receptor/mu-opioid receptor ligands: implications for therapeutic applications. J Pharmacol Exp Ther. 2009;331(3):954–64.
Ding H, Kiguchi N, Yasuda D, Daga PR, Polgar WE, Lu JJ, et al. A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Sci Transl Med. 2018;10(456).
Schunk S, Linz K, Hinze C, Frormann S, Oberbörsch S, Sundermann B, et al. Discovery of a potent analgesic NOP and opioid receptor agonist: cebranopadol. ACS Med Chem Lett. 2014;5(8):857–62.
Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M, et al. Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J Pharmacol Exp Ther. 2014;349(3):535–48.
Cami-Kobeci G, Polgar WE, Khroyan TV, Toll L, Husbands SM. Structural determinants of opioid and NOP receptor activity in derivatives of buprenorphine. J Med Chem. 2011;54(19):6531–7.
Kiguchi N, Ding H, Cami-Kobeci G, Sukhtankar DD, Czoty PW, DeLoid HB, et al. BU10038 as a safe opioid analgesic with fewer side-effects after systemic and intrathecal administration in primates. Br J Anaesth. 2019;122(6):e146–e56.
Zaveri NT, Jiang F, Olsen C, Polgar WE, Toll L. Designing bifunctional NOP receptor-mu opioid receptor ligands from NOP receptor-selective scaffolds. Part 1. Bioorg Med Chem Lett. 2013;23(11):3308–13.
Journigan VB, Polgar WE, Khroyan TV, Zaveri NT. Designing bifunctional NOP receptor-mu opioid receptor ligands from NOP-receptor selective scaffolds. Part II. Bioorg Med Chem. 2014;22(8):2508–16.
Zaveri NT, Yasuda D, Journigan VB, Daga PD, Jiang F, Olsen C. Structure-activity relationships of Nociceptin Receptor (NOP) Ligands and the Design of Bifunctional NOP/mu opioid receptor-targeted Ligands. In: Ko MC, Husbands SM, editors. Research and Development of Opioid-Related Analgesics: American Chemical Society; 2013. p. 145–60.
Zaveri NT, Meyer ME. NOP-Targeted Nonpeptide Ligands. Handb Exp Pharmacol. 2019;254:37–67.
Kamakolanu UG, Meyer ME, Yasuda D, Polgar WE, Marti M, Mercatelli D, et al. Discovery and structure-activity relationships of nociceptin receptor partial agonists that afford symptom ablation in Parkinson's disease models. J Med Chem. 2020;63(5):2688–704.
Daga PR, Zaveri NT. Homology modeling and molecular dynamics simulations of the active state of the nociceptin receptor reveal new insights into agonist binding and activation. Proteins. 2012;80(8):1948–61.
Zaveri N, Jiang F, Olsen C, Polgar W, Toll L. Small-molecule agonists and antagonists of the opioid receptor-like receptor (ORL1, NOP): ligand-based analysis of structural factors influencing intrinsic activity at NOP. AAPS J. 2005;7(2):E345–52.
Meng F, Taylor LP, Hoversten MT, Ueda Y, Ardati A, Reinscheid RK, et al. Moving from the orphanin FQ receptor to an opioid receptor using four point mutations. J Biol Chem. 1996;271(50):32016–20.
Meng F, Ueda Y, Hoversten MT, Taylor LP, Reinscheid RK, Monsma FJ, et al. Creating a functional opioid alkaloid binding site in the orphanin FQ receptor through site-directed mutagenesis. Mol Pharmacol. 1998;53(4):772–7.
Lipiński PFJ, Kosson P, Matalińska J, Roszkowski P, Czarnocki Z, Jarończyk M, et al. Fentanyl family at the Mu-opioid receptor: Uniform Assessment of Binding and Computational Analysis. Molecules. 2019;24(4).
Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, et al. Structure of the μ-opioid receptor-G(i) protein complex. Nature. 2018;558(7711):547–52.
Zaveri NT, Marquez PV, Meyer ME, Polgar WE, Hamid A, Lutfy K. A novel and selective nociceptin receptor (NOP) agonist (1-(1-((cis)-4-isopropylcyclohexyl) piperidin-4-yl)-1H-indol-2-yl)methanol (AT-312) decreases acquisition of ethanol-induced conditioned place preference in mice. Alcohol Clin Exp Res. 2018;42:461–71.
Zaveri NT, Marquez PV, Meyer ME, Hamid A, Lutfy K. The nociceptin Receptor (NOP) agonist AT-312 blocks acquisition of morphine- and cocaine-induced conditioned place preference in mice. Front Psychiatry. 2018;9:638.
Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485(7398):395–9.
Miller RL, Thompson AA, Trapella C, Guerrini R, Malfacini D, Patel N, et al. The importance of ligand-receptor conformational pairs in stabilization: spotlight on the N/OFQ G protein-coupled receptor. Structure. 2015;23(12):2291–9.
Acknowledgements
This work was supported by grants R01DA027811 (N.T.Z.), R43NS070664 (N.T.Z.), R44DA042465 (N.T.Z) and NIAAA Contracts HHSN275201300005C and HHSN275201500005C from the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
M.E.M and A.D. contributed equally to this work. M.E.M, A.D., and D.Y. conducted the design, chemical synthesis, and SAR analysis of the NOP ligands. A.D. conducted the molecular docking experiments. N.T.Z conducted the design and the SAR and data analysis, and supervised the study.
Corresponding author
Additional information
Guest Editors: Diane Burgess, Marilyn Morris and Meena Subramanyam
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 3418 kb)
Rights and permissions
About this article

Cite this article
Meyer, M.E., Doshi, A., Yasuda, D. et al. Structure-Based SAR in the Design of Selective or Bifunctional Nociceptin (NOP) Receptor Agonists. AAPS J 23, 68 (2021). https://doi.org/10.1208/s12248-021-00589-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-021-00589-7