
Abstract
Immune checkpoint inhibitors (ICPI) have revolutionized cancer therapy and provided clinical benefit to thousands of patients. Despite durable responses in many tumor types, the majority of patients either fail to respond at all or develop resistance to the ICPI. Furthermore, ICPI treatment can be accompanied by serious adverse effects. There is an urgent need for identification of patient populations that will benefit from ICPI as single agents and when used in combinations. As ICPI have achieved regulatory approvals, accompanying biomarkers including PD-L1 immunohistochemistry (IHC) and tumor mutational burden (TMB) have also received approvals for some indications. The ICPI pembrolizumab was the first example of a tissue-agnostic FDA approval based on tumor microsatellite instability (MSI)/deficient mismatch repair (dMMR) biomarker status, rather than on tumor histology assessment. Several other ICPI-associated biomarkers are in the exploratory stage, including quantification of tumor-infiltrating lymphocytes (TILs), gene expression profiling (GEP) of an inflamed microenvironment, and neoantigen prediction. TMB and PD-L1 expression can predict a subset of responses, but they fail to predict all responses to checkpoint blockade. While a single biomarker is currently limited in its ability to fully capture the complexity of the tumor-immune microenvironment, a combination of biomarkers is emerging as a method to improve predictive power. Here we review the steadily growing impact of comprehensive genomic profiling (CGP) for development and utilization of predictive biomarkers by simultaneously capturing TMB, MSI, and the status of genomic targets that confer sensitivity or resistance to immunotherapy, as well as detecting inflammation through RNA expression signatures.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent advances in cancer biology and tumor immunology have ushered in a new era of cancer immunotherapy in the form of immune checkpoint inhibitors (ICPI) (1). Modern immunotherapy has transformed the practice of medical oncology. A recent study found that the estimated percentage of US patients with cancer who are eligible for ICPI increased from 1.54% in 2011 to 43.63% in 2018 (2). The efficacy of ICPI treatment, whether alone or in various combinations is reduced by primary or acquired resistance to treatment, and the impact of ICPI on overall survival (OS) of cancer patients remains limited. Initially, the percentage of patients estimated to respond to ICPI was 0.14% in 2011 and increased to 12.46% in 2018 (2). ICPI use may also cause unique side effects unlike those associated with targeted agents and chemotherapies, resulting from the immune system targeting normal immune cells as well. The most common adverse effects encountered are colitis, hepatitis, adrenocorticotrophic hormone insufficiency, hypothyroidism, type 1 diabetes, acute kidney injury, and myocarditis (3). With a main goal of expanding immunotherapy applications, the identification and utilization of biomarkers to guide both positive and negative predictive decision insights are essential. Important positive impact measures include the accurate prediction of initial and long-term durable therapeutic responses and improvement in quality of life, while the identification of negative prediction impact measures, such as predisposition to ICPI adverse effects, will increase cost-effectiveness, due to decreased ER utilization and hospitalizations.
Current FDA-approved predictive biomarkers linked to ICPI use include PD-L1 expression via immunohistochemistry (IHC) and tumor mutational burden (TMB) detected using comprehensive genomic profiling (CGP), with the identification of microsatellite instability (MSI)/mismatch repair deficiency (dMMR) by multiple modalities as part of an FDA-approved indication for pembrolizumab. However, when the significant frequency of non-responders and the substantial costs of the typical treatment using ICPI of greater than $150,000 per year are considered (4), it is apparent that patient selection using additional validated biomarkers is needed to optimize clinical outcomes and healthcare resource utilization. In this review, we will explore the current landscape of biomarkers to predict response to ICPI, their current successes and limitations, and emerging novel biomarkers that have potential to further improve clinical efficacy.
Immune Checkpoint Blockade
In 2013, Chen and Mellman proposed the cancer immunity cycle as a framework to explain how genomic alterations inherent to every cancer initiate a complex multi-step process that culminates with an anti-tumor immune response as illustrated in Fig. 1 (5,6). Today, this model forms the scientific basis for application of immune checkpoint blockade in the clinic, which is the foundation of modern immunotherapy.

Framework for signaling between activated T cells, antigen presenting cells, and tumor cells. T cell receptor (TCR) interacts with neoantigen/major histocompatibility complex (MHC) to induce a stimulatory signal. T cells are kept in check by activation of various coinhibitory receptors as outlined in the diagram. When these receptors or ligands are blocked by therapeutic antibodies, ensuing T cell activation causes release of cytotoxic IFN-γ, perforin, and granzyme. IFN-γ activates the interferon pathway through IFNGR1/2, JAK1/2 and STAT1
Since the first approval of anti-CTLA-4 treatment for patients with metastatic melanoma in 2011, a series of fast-paced ICPI approvals have changed how advanced cancer is treated. At least seven monoclonal antibodies have gained US regulatory approval in 45 different indications targeting the CTLA-4 and PD-1/PD-L1 axes (7). However, with few exceptions such as Hodgkin’s lymphoma and Merkel cell carcinoma, the overall response rate (ORR) to ICPI monotherapy remains in the 10–30% range (8,9), prompting an exhaustive search to understand the biology of primary and acquired resistance to ICPI, and define better predictive biomarkers and novel combination regimens. In a manner analogous to the activation of parallel signaling pathways that often mediate acquired resistance to targeted therapy in oncogene-addicted cancers, at least four novel inhibitory receptors (LAG-3, TIM3, TIGIT, and VISTA) are often upregulated in ICPI-resistant cancers and are currently being explored as targets in conjunction with anti-PD-1/PD-L1 or anti-CTLA-4 (10).
As the widespread use of ICPI continues to expand, the identification and validation of biomarkers capable of predicting both ICPI responses and adverse events have never been more important.
Biomarkers for Immune Checkpoint Inhibitors
The response to ICPI is influenced by both tumor intrinsic and extrinsic factors, which can be monitored using various biomarkers (Fig. 2). These biomarkers can be detected using a wide array of molecular techniques, including detection of PD-L1 expression by immunohistochemistry (IHC), degree of T cell infiltrates within the tumor tissue by IHC or immunofluorescence, as well as genomic/ transcriptomic biomarkers such as MSI, TMB, signaling alterations, and IFN-γ pathway activity using CGP. CGP is a next-generation sequencing (NGS)-based methodology that simultaneously detects the four main classes of cancer-associated genomic alterations and generates genomic signatures, designed to provide prognostic, diagnostic, and predictive insights that inform treatment decisions for individual patients across all cancer types.

Overview of established and experimental immunotherapy biomarkers. PD-L1 expression, TMB value, and MSI status have been FDA approved as biomarkers for ICPI therapy under specific conditions. Oncogenic signaling, which is analyzable through CGP, can modulate the response to checkpoint inhibitors. Efforts to incorporate TILs and inflamed RNA expression signatures into composite biomarkers are ongoing
PD-L1 Immunohistochemistry as the First FDA-Approved Biomarker for Immune Checkpoint Inhibitors
PD-L1 is an inducible type-I transmembrane ligand expressed on a subset of macrophages, T-, B-lymphocytes, NK cells, and endothelial cells in an inflammatory microenvironment, where it maintains host immune response and prevents autoimmunity by binding to the PD-1 receptor and inducing an immune checkpoint (11,12,13). This function has been usurped by tumor cells in an effort to evade immune surveillance and forms the basis for the adoption of PD-L1/PD-1 blockade as an anti-cancer therapy (12,14,15). Upregulation of PD-L1 through inflammatory signaling is mediated through IFN-γ (16,17) produced by T-cells and NK cells. IFN-γ signals through the JAK-STAT pathway and activates several interferon-responsive factors (IRF) of which IRF-1 is responsible for PD-L1 upregulation (18). Given the critical role played by IFN-γ in upregulating PD-L1, expression of PD-L1 detected by IHC is often viewed as a surrogate marker of an inflamed tumor microenvironment and enhanced IFN-γ activity.
The most well-studied candidate biomarker in this field and first to be granted regulatory approval as a companion diagnostic test for PD-1 blockade was PD-L1 protein expression detected by IHC (19). PD-L1 expression varies significantly among different tumor types, and this difference generally correlates with response to PD-1 pathway blockade (20,21), thus providing both a mechanistic explanation for how tumors evade host immune response (22) and a rationale for the development of PD-L1 expression as a response biomarker (23). Data from these early phase clinical trials provided the scientific rationale for PD-L1 IHC testing and suggested that expression of PD-L1 predicts a favorable response to ICPI (24). This has led to the development of multiple companion diagnostic tests to accompany PD-1/PD-L1 regulatory approvals that have included assessment of PD-L1 expression on tumor cells, intra-tumoral inflammatory cells, or both (25). Expression of PD-L1 on immune cells is important in mediating immunosuppressive activity (26) and predicting clinical response to ICPI based on the numerous PD-L1 IHC companion diagnostic (CDx) approvals in the past few years that is dependent solely on immune cells expression or a combination of immune cell and tumor cell expression of PD-L1. For example, PD-L1 is expressed on antigen presenting cells (APC), where a cis interaction with CD80 is required for optimal immune responses (27).
The initial approval of pembrolizumab for previously treated non-small cell lung cancer (NSCLC) patients was based on the KEYNOTE-001 trial and PD-L1 expression level of ≥ 50% on tumor cells (tumor proportion score (TPS) ≥ 50%) using the DAKO 22C3 pharmDx assay (23). Subsequent trials lowered that threshold to ≥ 1% based on KEYNOTE-010 (28). Pembrolizumab approval for treatment-naïve NSCLC patients followed a similar route where initial approval based on KEYNOTE-024 required PD-L1 TPS ≥ 50%, and this was subsequently lowered to TPS ≥ 1% based on KEYNOTE-042. This variable threshold for the definition of a positive cut-off among treatment-naïve patients and those previously treated sets immune therapy biomarkers apart from targeted therapy, where typically a binary system exists for what constitutes a positive threshold for treatment selection. Approvals for nivolumab and atezolizumab in previously treated NSCLC patients were granted independent of PD-L1 expression.
The Blueprint project compared four trial-validated anti-PD-L1 IHC assays in 39 NSCLC tumor samples (Table I). The study showed that the percentage of PD-L1 stained tumor cells was comparable between three of the four assays (22C3, 28–8 and SP263) but consistently lower with the fourth assay (SP142). All the assays showed greater variability in PD-L1 staining on immune cells than on tumor cells. The study indicated that interchanging assay methods and cut-off values for PD-L1 positivity can lead to inconsistent classifications of PD-L1 status in some patients, highlighting the challenges of the current IHC approach to assessing PD-L1 protein expression.
In addition to the detection of PD-L1 protein expression using IHC, CGP can be used for detection of amplification of the chromosome 9p24.1 locus encompassing the genes encoding PD-L1 (CD274), PD-L2, and JAK2 (Fig. 3) (29), which has been observed in several malignancies including Hodgkin’s lymphoma, various B cell lymphomas, as well as in solid tumors. The CD274 gene amplification is quite uncommon (< 2% of solid tumors), but, when it occurs, is nearly always associated with high PD-L1 IHC staining. Preliminary data indicate that amplification is associated with high response rates to ICPI (29).

Copy number plot showing amplification of CD274 in metastatic breast cancer. This tumor from a 64-year-old woman was MSS and featured a TMB of 4 mutations/Mb. Genomic findings included CD274 (PD-L1) amplification at 25 copies/cell, as indicated by the arrow
Taken together, PD-L1 is a sub-optimal predictive biomarker for patients that may derive benefit from ICPI treatment. Underscoring this point, patients harboring tumors that are negative, or < 1% positive, for PD-L1 often still experience impactful clinical responses from ICPI (30,31,32,33,34). Thus, additional biomarkers are urgently needed to enhance the predictive power of PD-L1 IHC assays. To provide an overview, all the immune checkpoint biomarkers that will be discussed are listed in Table II.
Mismatch Repair Deficiency/ Microsatellite Instability Is a Powerful Predictor of Sensitivity to Immune Checkpoint Inhibitors
In 2017, the FDA granted a landmark first tissue agnostic approval of pembrolizumab based on tumor biomarker status only for the treatment of patients harboring microsatellite instability high (MSI-H) or mismatch repair deficient (dMMR) unresectable/metastatic solid tumors (35,36). While MSI-H cases occur throughout the tumor spectrum at low frequency, they are most prevalent in colorectal (CRC) and endometrial carcinomas at approximately 15% frequency (5% in metastatic CRC) (37,38).
To determine MSI/MMR status, four MMR proteins, MSH2, MSH6, MLH1, and PMS2 are typically first assessed in tumor tissues using IHC (39). If one or more of these proteins are not detected through IHC, the CRC is classified as mismatch repair deficient (dMMR). Due to errors in the function of MMR during DNA replication, MSI can result. While MSI-H status is associated with dMMR, differences in how MSI and MMR are measured translate into less than 100% congruent results, for example, due to monoallelic loss or loss of another uncharacterized MMR gene or promoter methylation (40). Early testing of MSI was performed on only a few canonical microsatellite loci, BAT26, BAT26, D5S346, D2S123, and D17S250 (41) via PCR followed by capillary electrophoresis, a method requiring the analysis of paired normal/tumor specimens. Additional MSI detection methods are currently available, including single-molecule molecular inversion probes (smMIPs), droplet-digital PCR, and next-generation sequencing (NGS) (42,43). NGS-based methodologies and computational algorithms have allowed unbiased, genome-wide screening of the molecular fingerprints of MSI, greatly increasing the sensitivity of MSI detection (44). A novel method of calling MSI using a targeted hybrid capture NGS-approach was recently shown to be 97% concordant with current standards, PCR and IHC, exhibiting 95% sensitivity and 98% specificity, and unlike many PCR-based tests, it does not require a matched normal tissue (40). A recent study compared MSI status assessed by NGS or PCR fragment analysis in 2189 matched cases from 26 cancer types and found the sensitivity of the NGS test to be 95.8% and specificity 99.4% relative to PCR (45). Recently, different methods were compared for assessment of MSI status, including PCR, NGS, and IHC (46).
The analytical and clinical validation of assays is essential prior to use in clinical trials and patient care. The Centers for Medicare & Medicaid Services (CMS) regulates all clinical laboratory testing in the USA through the Clinical Laboratory Improvement Amendments (CLIA) for Laboratory Developed Tests with best practice guidelines for NGS-based genomic testing outlined by the Association of Molecular Pathology with liaison representation from the College of American Pathologists (47). Even more robust performance requirements from the US FDA need to be demonstrated prior to approval or clearance of a diagnostic device (48). Foundation Medicine’s FoundationOne®CDx is currently FDA approved and Memorial Sloan Kettering Cancer Center’s MSK-IMPACT is FDA authorized for the detection of MSI in tumor tissue using CGP. In this exceptional case, the FDA granted approval of pembrolizumab for treatment of MSI-H/dMMR cancers without an accompanying CDx, because the clear benefits from use of the product were viewed as outweighing the risks from lack of an approved CDx (35). MSI screening, when integrated into CGP, can routinely be applied broadly across many tumor types, where PCR would be impractical due to low MSI frequencies. The utilization of CGP for the detection of MSI also allows the detection of alterations in other potentially targetable genes to inform precision treatment options, in addition to identifying alterations within the MLH1, MSH2, MSH6, PMS2, and EPCAM genes found in patients with Lynch Syndrome, an inherited condition that causes an increased risk for CRC, endometrial, and other cancers. As a major advance for patients where the amount of tissue for IHC is insufficient, MSI can also be detected by ctDNA-based liquid biopsy using hybrid capture gene panels that also include targeted microsatellite loci (49,50,51). By overcoming major challenges such as repetitive loci, low tumor fraction, high-level technical noise through selection of informative microsatellite loci, and application of bioinformatics to improve sequencing accuracy, this important test is now ready for further validation in prospective studies.
Neoantigens and Tumor Mutational Burden
Cancer develops when the repair of DNA damage sustained during exposure to exogenous and endogenous carcinogens gives rise to genomic alterations that disrupt the function of oncogenes and tumor suppressors and allows the developing cancer cells to escape host surveillance. Neoantigens are defined as tumor-specific antigens that arise from non-synonymous mutations and other genetic alterations (52). These neoantigens, when processed into short peptides and presented by MHC molecules to T cells, stimulate the immune system recognition of cancer cells as “non-self” and enable subsequent immune-mediated attack (53). This nascent anti-tumor immune response is further amplified by immune checkpoint blockade (54,55). The number of mutations, and particularly the number of clonal immunogenic mutations, predicts tumor response to anti-CTLA4 and anti-PD-1 (56).
CGP has the ability to identify acquired non-germline mutations that have the potential to generate neoantigens, which in turn may be used to predict responses to ICPI (52,57). In contrast with measuring TMB directly, computational algorithms for neoantigen prediction from sequencing data have been developed and are constantly being refined, but current algorithms are not strongly predictive of ICPI responses (52,58). However, efforts to use HLA genotyping to predict the relative efficiency of neoantigen presentation, and the use of this information together with TMB to predict ICPI responses is showing promise (59,60).
Determining TMB from sequencing data has emerged as a major predictive biomarker for ICPI responses (61,62,63,64), especially in ICPI monotherapy trials. TMB is a measure of the number of somatic, non-driver mutations per Mb of DNA, which includes substitutions, insertions, and deletions (indels) (61,62,63,64). TMB is often considered a surrogate for neoantigen load. Mutational processes, such as those derived from cigarette smoke (lung and bladder cancer) and ultraviolet light (melanoma), are known to increase TMB, and the concomitant higher mutational burden leads to an increase in neoantigens and immunogenicity, thus providing a rationale for the role of TMB as predictive biomarker for ICPI response. The range of somatic mutations among tumors varies from 0.01 per Mb to more than 400 mutations per Mb (65,66). TMB was originally assessed by whole exome sequencing (WES); however the impracticality of applying WES on a routine clinical scale has refocused the application of TMB towards CGP panels (66). To date, Foundation Medicine’s FoundationOne®CDx assay, in which TMB is calculated based on a ~ 0.8 Mb genomic region, is the only FDA approved assay which includes TMB as part of its tumor profiling claim. Two additional assays have received FDA authorization, including MSK-IMPACT, which covers ~1.5 Mb over 468 genes and Omics Core from NantHealth, which covers ~39 Mb over 19,396 genes.
Differences in panel size, technical sensitivity of the assay, pre-analytical and analytical variables, in addition to the underlying bioinformatics pipelines, however, are known causes of variability in TMB estimates across laboratories (62,64,67). Size of the genomic panel has been identified as a key indicator in the sensitivity of panels to capture accurate TMB measurements. In general, evidence suggests that gene panels of ≥ 0.5 Mb are needed to accurately stratify TMB subpopulations down to levels of 10 mut/Mb (66). Beyond the minimum size of the genomic content to accurately calculate TMB, cancer panels by design over-represent cancer-related genes and thus filtering the driver mutations from the calculation is important to reduce the bias that can lead to TMB over-estimations. Furthermore, TMB is a continuous variable that has been forced into a binary definition of high or low based on discrete cutoffs. More work is needed to understand the relationship of clinical response along the continuum of TMB scores and how discrete values can provide more clinical information beyond a simple dichotomous classification. Additionally, harmonization efforts are currently underway to ensure alignment and improve interchangeability between TMB estimates generated from different targeted gene panels or WES (64,68,69,70), an activity essential to ensure this biomarker provides consistent information to inform treatment decisions across diagnostic platforms. Furthermore, studies indicate that TMB acts as an independent biomarker from PD-L1 with limited overlap (Fig. 4) (71). While TMB may outperform PD-L1 as a predictive biomarker, the use of both tests may enhance the identification of patients that might benefit from ICPI (72,73).

The relationship between PD-L1 IHC, tumor mutation burden, and microsatellite instability across multiple tumor types with a PD-L1 IHC CDx. Data was available for all three biomarkers for 22,592 clinical cases in multiple tumor indications with PD-L1 IHC CDx. Tumor types in this cohort stained with a PD-L1 IHC CDx assay and interpreted with associated cut-offs included: non-small cell lung cancer (TPS ≥ 1), esophagus squamous cell carcinoma (CPS ≥ 10), urothelial carcinoma (CPS ≥ 10), cervix cancer (CPS ≥ 1), gastric/gastroesophageal adenocarcinoma (CSP ≥ 1), head and neck squamous cell carcinoma (CPS ≥ 1) stained with the DAKO PD-L1 22C3 CDx assay; and breast carcinoma (IC ≥ 1) stained with VENTANA SP142 CDx assay. TMB and MSI were determined using the FoundationOne®CDx or FoundationOne® CGP assay. TMB+ was considered positive at the pan-tumor CDx cut-off ≥ 10 mutations/Mb and MSI+ was equivalent to MSI-High
MSI-H/dMMR tumors are almost always accompanied by a high TMB, ranging from as low as 6 mutations/Mb to as high as 819 mutations/Mb in MSI-H CRC (74). In contrast, most tumors with high TMB are not MSI-H. Tumors with exceptionally high TMB respond favorably to ICPI such as pembrolizumab. MSI-H CRC tumor class can be subdivided according to the TMB value. Specifically, the MSI-H population with > 37 mutations/Mb predicted a positive impact on tumor response, progression-free survival (PFS), and overall survival (74). Furthermore, while the majority of microsatellite stable (MSS) colorectal tumors are predicted to be resistant to ICPI, a minority of about 3% has high TMB and may respond favorably (75). This observation may increase the number of colorectal tumors eligible for ICPI by 50% beyond the MSI-H population (75). Mutations in the replicative polymerases POLE and POLD are also associated with ultra-high TMB values, and case studies suggest favorable responses to checkpoint blockade (76,77). As a specific example, a patient with a rare case of hypermutated, MSS castration-resistant prostate cancer was found by CGP to harbor a POLE V411L mutation, and the patient is demonstrating a long-term complete response (> 6 years) to ICPI (78).
Several large-scale clinical studies have evaluated the relationship between TMB and treatment outcome in cancer patients and found notable clinical benefit associated with the TMB high population especially in single ICPI treatment protocols. A few of the landmark studies merit further discussion. In a retrospective study of melanoma, Snyder et al. found that higher TMB was correlated with overall survival from ipilimumab or tremelimumab treatment (79). Rizvi et al., in a prospective study of NSCLC, found that a TMB greater than the median was associated with longer PFS and durable responses using pembrolizumab (80). Van Allen et al. performed WES from 110 patients with melanoma treated with ipilimumab and demonstrated that mutational load, neoantigen load, and expression of cytolytic markers were associated with clinical benefit from ipilimumab (81). Chalmers et al. established potentially broad impact of TMB for prediction of ICPI responses in a range of tumor types by analyzing 100,000 human cancer genomes (66). Goodman et al. performed a retrospective pan-tumor study and demonstrated higher response rate and increased PFS when TMB ≥ 20 mutations/Mb after treatment with various ICPI (82). Yarchoan et al. showed correlation between clinical response to anti-PD-1/PD-L1 and TMB across 27 different tumor types (83). Similarly, Samstein et al. found that high TMB, calculated from MSK-IMPACT NGS, predicted better overall survival across multiple cancer types (57). In the CheckMate-227 trial for NSCLC, TMB was found to be predictive of PFS for nivolumab/ipilimumab treatment (32,84). In addition, a study of real-world cancer data from a clinical-genomic database revealed that TMB ≥ 20 mut/ Mb was associated with improved overall survival in NSCLC treated with ICPI (85). Collectively, these examples as well as many additional studies found an association between high TMB and favorable clinical response rates with a single immunotherapy agent (86).
In June 2020, FDA granted regulatory approval for the use of FoundationOne®CDx as the first companion diagnostic for pembrolizumab, Merck’s anti-PD-1 therapy, to identify patients with unresectable or metastatic TMB-H (≥ 10 mutations/Mb) solid tumors that that have progressed following prior treatment and who have no satisfactory alternative treatment options. This approval was based on a prospectively planned retrospective analysis of the KEYNOTE-158 open-label trial, which used a clinical trial assay (CTA) based on FoundationOne®CDx to determine TMB status in patient’s tumor tissue. The results showed that patients with TMB ≥ 10 mutations/Mb who were treated with pembrolizumab had a higher overall response rate (28%) compared with patients with TMB values < 10 mutations/Mb (7%) (87). As illustrated in Fig. 5, TMB-H has been detected in a large number of solid tumor types; therefore, this new approval may provide additional treatment options to a large number of advanced cancer patients. While TMB as a biomarker predictive of ICPI efficacy appears established for single agent trials, the predictive power of TMB for patients receiving combinations of ICPI with chemo is not as well-established (86).

The percentage of cases with TMB ≥ 10 Mut/Mb across various solid tumor types determined using the FoundationOne® CDx assay
For many patients, obtaining sufficient tissue for CGP can be a challenge, so the ability to generate predictive genomic data utilizing ctDNA from liquid biopsies would provide significant benefit to cancer patients. Additional benefits include ease of blood sample collection at one or multiple points and speed of processing (88). The analysis of ctDNA, however, presents considerable challenges for laboratories, and as in the case of tumor tissue, the entire liquid biopsy workflow needs to be carefully optimized, with appropriate quality controls included for the measurement of extraction efficiency and fragment size and assays appropriately validated for accuracy, reliability, and robustness (89,90,91). Several ctDNA companion diagnostics are being developed to predict and monitor responses to therapy, as well as detect emerging resistance. Currently, plasma-based measurement of TMB (bTMB) based on ctDNA is being evaluated as a non-invasive, predictive biomarker for ICPI efficacy. Gandara et al. published the clinical utility of a bTMB assay in a retrospective study of the POPLAR and OAK randomized NSCLC clinical trials, using the former as a training set and the latter for validation (92). By setting a cutoff for bTMB at 14 mutations/Mb, they were able to predict which patients would derive clinical benefit from atezolizumab. Furthermore, TMB values derived from plasma samples were generally correlated with TMB values obtained from tissue from the same patients; however many samples were discordant due to low tissue tumor purity or low cell-sfree DNA inputs. The BF1RST trial also supported the predictive value of bTMB in NSCLC (93). A study of the MYSTIC trial using GuardantOmni from Guardant Health also found bTMB to be predictive of PFS and OS benefit with durvalumab ± tremelimumab in NSCLC (94). In addition to NSCLC, a recent phase III EAGLE study demonstrated that bTMB can predict survival in head and neck squamous cell carcinomas treated with durvalumab + tremelimumab when compared with chemotherapy (95). Collectively, while these data suggest bTMB may be an actionable biomarker in NSCLC for ICPI therapy, additional prospective studies are needed to demonstrate clinical utility. FDA recently approved FoundationOne®Liquid CDx, which is a comprehensive liquid biopsy test with multiple CDx indications and offers pan-tumor profiling of over 300 genes (link to FDA label (96)); on the professional services page, bTMB is reported to support clinical use in cancer patients as deemed appropriate by the treating physician.
The Tumor Microenvironment Modulates T-Cell Infiltration and Responses to Checkpoint Inhibitors
The tumor microenvironment (TME) encompasses a broad range of cell types such as cancer-associated fibroblasts, endothelial cells, blood vessels, lymph vessels and a highly heterogeneous immune cell population (including T cells, B cells, NK cells, dendritic cells, monocytes, neutrophils, basophils, eosinophils) that coexist with and occasionally support the tumor cells (97). The complexity of the TME and the dynamic nature of the tumor-immune interplay makes predictions of ICPI responses challenging. ICPI are believed to act in large part by awakening pre-existing tumor immune responses, and effector T cells are critical for these responses. Therefore, immune cell infiltration of the tumor is a contributing factor to ICPI outcome. Specifically, the density of tumor-infiltrating lymphocytes (TILs) is a positive prognostic indicator. Tumors are classified according to four immune phenotypes as immune inflamed (previously referred to as “hot”), immune excluded, immunosuppressed, or immune desert (previously referred to as “cold”) (98). The immunoscore was devised to quantify the density of CD3+ and CD8+ T cells at the center and invasive margin of the tumor using IHC coupled with automated quantitative imaging (99,100). While the immunoscore was shown to have significant prognostic value in colon cancer, its potential value more broadly as a predictive marker for ICPI is still under evaluation (101).
Several transcriptomic signatures have been developed to monitor the TME and help to assess ICPI sensitivity or resistance. Ayers et al. compiled a NanoString 18-gene T cell-inflamed gene expression profile (GEP, also referred to a “Tumor Inflammation Signature” (TIS)) based on IFNγ responsive genes that appears to have predictive power for checkpoint therapy in various tumor types (102). Another gene expression signature, “immune-predictive score” (IMPRES), was built on a total of 297 patient samples derived from 10 datasets of patients treated with ICPI (103). The predictive power of IMPRES in melanoma was superior to other markers but remains to be tested in other tumor types. Jiang et al. developed the tumor immune dysfunction and exclusion (TIDE) RNA expression signature to model T cell dysfunction and exclusion, which are major causes of tumor immune evasion (104). TIDE was able to predict outcomes from treatment of melanoma patients with ICPI, as well as help to identify specific resistance mechanisms. It should be emphasized that all the above transcription signatures are derived from bulk RNA sequencing and therefore represent a composite picture of signaling from tumor cells, immune cells, and other cell types in the TME. Recently, single-cell RNA sequencing was leveraged to reveal resistance programs in melanoma that are associated with T cell exclusion and immune evasion, and this program could predict ICPI responses in multiple cohorts (105). There are ongoing efforts to test all these transcriptional signatures in a broader context of tumor types and in prospective clinical trials.
Multimodal Biomarkers Improve Prediction of Immune Checkpoint Responses
Over the past 5 years, the search for predictive immune oncology biomarkers to help guide patient selection for ICPI therapy has expanded from the relative simplicity of PD-L1 IHC to include TMB, MSI, GEP, multiplex IHC/immunofluorescence (mIHC/IF), as well as multimodality biomarker strategies that combine two or more of the above biomarkers. A recent meta-analysis comparing these four testing modalities evaluated specimens from 8135 patients with more than 10 different solid tumors (106). The authors assessed output of these different biomarkers as area under the curve (AUC) of the summary receiver operating characteristic (sROC) curves and concluded that TMB, PD-L1 IHC, and GEP demonstrated comparable efficacy in predicting response to anti–PD-1/PD-L1 treatment. However, mIHC/ IF and some biomarker combinations were associated with improved performance over PD-L1 IHC, TMB, or GEP alone, suggesting that mIHC/IF has diagnostic accuracy comparable with multimodal approaches in predicting response to anti–PD-1/PD-L1. Other studies have combined biomarkers as well. By integrating multiple clinical trials with genomic and biomarker databases, Cristescu et al. demonstrated that combining TMB together with GEP had a greater predictive power to stratify patients that will respond to ICPI (107). Miao et al. investigated markers beyond TMB and revealed the role of mutational clonality (representing intratumoral heterogeneity), mutational signatures, and genetic driver events in responses to immune checkpoint blockade (108). Anagnostou et al. demonstrated that tumor purity significantly impacts the predictive value of TMB and generated a multivariate model based on weighted contributions from TMB, tumor purity, a smoking signature, receptor tyrosine kinase (RTK) mutations, and HLA genetic variation (109). These combined biomarker approaches are particularly relevant in view of the expanding repertoire of candidate checkpoint inhibitors entering clinical trials in combination with anti-PD-1/PD-L1 (LAG3, TIM3, TIGIT, VISTA, GITR) (10). Matching individual patients/tumors to the correct checkpoint combination will require insight into the immune composition of TME in order to speed up trial accrual and subsequent FDA approval. Similarly, an understanding of various oncogenic signaling pathways and co-occurring mutations in tumor suppressors will be critical to help guide the pairing of small molecule inhibitors of PARP, BRAF, MEK, phosphatidylinositol 3-kinase (PI3K), and RTKs with checkpoint inhibitors. CGP provides enormous predictive value for ICPI responses by simultaneously reporting TMB value, MSI status, and oncogene and tumor suppressor alterations, while also allowing the identification of alterations that will guide these future combination therapy strategies (for example with BRAF, RTK or PI3K inhibitors). The ability of biomarkers to predict efficacy of ICPI must be evaluated for both single-agent ICPI regimens and in combination with other therapies.
Factors that Impact Response to Immune Checkpoint Blockade
Although durable responses to checkpoint blockade have been observed in a wide range of tumor types, most tumors fail to respond or eventually develop resistance. Both primary and acquired resistance is widespread, and it can be difficult to distinguish between these. ICPI responses have been attributed to the genomic correlates of T cells, tumor cells, or the TME. Cytotoxic T cell function requires proper tumor infiltration, effector function, reduced exhaustion, increased clonality, and enhanced stemness, properties which can be partially assessed using transcriptional signatures. Favorable responses to checkpoint blockade are correlated with high neoantigen load, increased TMB, upregulated PD-L1, and certain inactivating mutations in SERPIN3 and SERPIN4, which are associated with benefit and longer survival in melanoma (110). Also, a recent study of NSCLC patients demonstrated that mutations in the putative tumor suppressor FAT1 were a favorable predictive factor independent of TMB (111). The diagram in Fig. 6 provides an overview of potential resistance mechanisms for ICPI. One of the prevalent resistance mechanisms corresponds to deficient antigen presentation, which can occur on several levels. Mutations or loss of heterozygosity (LOH) of B2M have been shown to confer resistance to checkpoint blockade in melanoma and other tumors (112,113,114,115). HLA genes, which encode MHC I proteins that present intracellular peptides, have been shown to undergo LOH and thereby contribute to immune evasion for example in NSCLC (116). Conversely, HLA class I diversity positively impacts responses to checkpoint blockade (117). Antigen presentation also involves IFN-γ and the downstream JAK/ STAT pathway. Genomic defects linked to resistance have been detected in IFNγ receptor (IFNGR1), IFNGR2, IRF1, JAK1 and JAK2, as well as through amplification of the inhibitor SOCS1 (115,118,119,120).

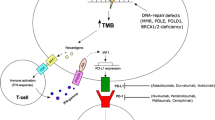
Landscape of immune checkpoint blockade resistance pathways. The diagram shows how tumor proteins are degraded through the proteasome to generate neoantigens that subsequently traffic through the ER and Golgi to finally be presented via MHC-I on the cell surface. As indicated by specific examples, every step of this process can be interfered with to prevent an anti-tumor immune response. Oncogenic signaling mechanisms known to modulate ICPI responses are also depicted. The predictive versus prognostic role of STK11 and KEAP1 in checkpoint responses remains debatable
Another major contribution to resistance comes from activation of oncogenic signaling pathways or loss of tumor suppressor function, both of which can frequently be detected by CGP. Mutational inactivation of the tumor suppressors KEAP1 and STK11/ LKB1, individually or in combination, in NSCLC was initially shown to act as a negative predictor of ICPI responses (121,122). Preserved STK11 function appears to play a role in normal lymphocyte trafficking. However, a recent study based on real-world data from the Foundation Medicine-Flatiron Health NSCLC Clinico-Genomic database (CGDB) suggests that mutations in KEAP1 and STK11 are prognostic rather than predictive, since they did not exhibit treatment-specific effects (123). Although STK11/KEAP1 mutations significantly reduce the likelihood of ICPI benefit in NSCLC, given the lack of other treatment options for advanced disease patients, ICPI might still be considered, even when these tumor suppressors are inactivated. Defects in the PTEN tumor suppressor were linked in melanoma to resistance to anti-PD-1/ PD-L1, mainly due to increased suppressive cytokines and reduced autophagy (124,125). Furthermore, mutational activation of the WNT/ β-catenin pathway in melanoma caused immune cell exclusion, reducing dendritic cell recruitment and T cell priming, thus resulting in poor outcome from ICPI (126). In NSCLC, EGFR mutations were associated with low response rate to ICPI and poor infiltration of CD8+ T cells relative to KRAS-driven tumors (127,128). The effect of KRAS mutations was variable, depending on tumor context and co-occurring mutations, whereas BRAF mutations were associated with favorable ICPI responses (129). Loss-of-function mutations of SWI/ SNF chromatin modifier genes such as PBRM1, ARID1A, and SMARCA4 have been linked with improved ICPI responses, but these observations require confirmation in additional studies. In the case of PBRM1 loss, the evidence has been confounding insofar as supporting contradictory roles in either resistance or susceptibility to checkpoint blockade, and uncertainty also remains whether this effect is associated with prior antiangiogenic anti-VEGF therapy (108,130,131,132,133). The reported association of ARID1A loss with impaired MMR and TMB-H may underlie favorable ICPI responses (134).
There is considerable concern that in a minority of cases ICPI treatment can lead to hyperprogressive disease (HPD), but the evidence is mixed on the specific biological underpinnings, and whether this is even a true phenomenon (135). A recent study of 214 patients with multiple tumors found HPD in 15% and an associated OS of 4.8 months versus 8.7 months for patients with or without HPD, respectively (136). Another study of 155 patients with various tumors reported that 4% of patients experienced HPD after anti-PD-1/ PD-L1 therapy, as defined by <2 month “time to treatment failure,” a tumor burden increase > 50% and greater than twofold tumor growth rate compared with pre-immunotherapy (137). Specifically, they demonstrated that 4/6 patients with tumors harboring MDM2/4 amplifications and 2/10 with EGFR mutations exhibited HPD. It remains unclear if HPD is associated specifically with ICPI or would occur regardless of the agent used. In another retrospective study further validating this data, the incidence of HPD was 66% for MDM2/4 and 50% for EGFR amplification; furthermore, they also found HPD in 43% of patients with 11q13 amplification (138). Taken together, while the numbers are small and prospective study validation is needed, these data suggest that patients with MDM2/4 amplification, EGFR activation, or 11q13 amplification may be at risk for HPD.
Conclusion
No single biomarker is likely to reliably predict responses to ICPI treatment across different tumor types and immunotherapy agents due to the complex and dynamic nature of tumor-immune cell interactions. The most highly characterized biomarkers to date, PD-L1 protein expression, and pan-tumor biomarkers MSI-H/dMMR and TMB, are important, but not optimal, due to inconsistencies related to tumor biology, protocols, and interpretations. Given the importance of the accurate and sensitive detection of these complex biomarkers, in addition to the large number of additional molecular biomarkers associated with ICPI resistance and response that can also be detected using CGP, this testing should be integrated into each cancer patient’s journey to ensure appropriate ICPI therapy utilization. Moving forward, future, even more complex composite biomarkers comprised of PD-L1 expression, TMB, inflammation signatures, TILs, and oncogenic alterations assessed by CGP are likely to be paramount for accurately predicting responses to immune checkpoint blockade. The development of algorithms to weigh the biomarkers into an ICPI Response Prediction Score could potentially emerge as a superior method of prediction of response to single ICPI or combined ICPI with chemotherapy or other treatment regimes.
References
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw Open. 2019;2(5):e192535.
Bajwa R, Cheema A, Khan T, Amirpour A, Paul A, Chaughtai S, et al. Adverse effects of immune checkpoint inhibitors (programmed Death-1 inhibitors and cytotoxic T-lymphocyte-associated Protein-4 inhibitors): results of a retrospective study. J Clin Med Res. 2019;11(4):225–36.
Galluzzi L, Chan TA, Kroemer G, Wolchok JD, Lopez-Soto A. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. 2018;10(459).
Blank CU, Haanen JB, Ribas A, Schumacher TN. Cancer immunology. The “cancer immunogram”. Science. 2016;352(6286):658–60.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer. 2019;7(1):278.
Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015;23:32–8.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Five-year survival and correlates among patients with advanced melanoma, renal cell carcinoma, or non–small cell lung Cancer treated with Nivolumab. JAMA Oncol. 2019;5(10):1411–20.
Andrews LP, Yano H, Vignali DA. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol. 2019:1–10.
Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–9.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34.
Mazanet MM, Hughes CC. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol. 2002;169(7):3581–8.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800.
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci. 2002;99(19):12293–7.
de Kleijn S, Langereis JD, Leentjens J, Kox M, Netea MG, Koenderman L, et al. IFN-γ-stimulated neutrophils suppress lymphocyte proliferation through expression of PD-L1. PLoS One. 2013;8(8).
Schoop R, Wahl P, Le Hir M, Heemann U, Wang M, Wüthrich RP. Suppressed T-cell activation by IFN-γ-induced expression of PD-L1 on renal tubular epithelial cells. Nephrol Dial Transplant. 2004;19(11):2713–20.
Lee S-J, Jang B-C, Lee S-W, Yang Y-I, Suh S-I, Park Y-M, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-γ-induced upregulation of B7-H1 (CD274). FEBS Lett. 2006;580(3):755–62.
Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–87.
Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–75.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–28.
Khunger M, Hernandez AV, Pasupuleti V, Rakshit S, Pennell NA, Stevenson J, et al. Programmed cell death 1 (PD-1) ligand (PD-L1) expression in solid tumors as a predictive biomarker of benefit from PD-1/PD-L1 axis inhibitors: a systematic review and meta-analysis. JCO Precis Oncol. 2017;1:1–15.
Hansen AR, Siu LL. PD-L1 testing in cancer: challenges in companion diagnostic development. JAMA Oncol. 2016;2(1):15–6.
Oh SA, Wu D-C, Cheung J, Navarro A, Xiong H, Cubas R, et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Can. 2020:1–11.
Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, et al. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science. 2019;364(6440):558–66.
Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–50.
Goodman AM, Piccioni D, Kato S, Boichard A, Wang HY, Frampton G, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 2018;4(9):1237–44.
Bhaijee F, Anders RA. PD-L1 expression as a predictive biomarker: is absence of proof the same as proof of absence? JAMA Oncol. 2016;2(1):54–5.
Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in advanced squamous-cell non-small-cell lung Cancer. N Engl J Med. 2015;373(2):123–35.
Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus Ipilimumab in advanced non-small-cell lung Cancer. N Engl J Med. 2019;381(21):2020–31.
Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–7.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–301.
Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: Pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res. 2019;25(13):3753–8.
Sidaway P. MSI-H: a truly agnostic biomarker? Nat Rev Clin Oncol. 2020;17(2):68.
Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res. 2016;22(4):813–20.
Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16(6):361–75.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073–87 e3.
Trabucco SE, Gowen K, Maund SL, Sanford E, Fabrizio DA, Hall MJ, et al. A novel next-generation sequencing approach to detecting microsatellite instability and Pan-tumor characterization of 1000 microsatellite instability-high cases in 67,000 patient samples. J Mol Diagn. 2019;21(6):1053–66.
Bacher JW, Flanagan LA, Smalley RL, Nassif NA, Burgart LJ, Halberg RB, et al. Development of a fluorescent multiplex assay for detection of MSI-high tumors. Dis Markers. 2004;20(4–5):237–50.
Li K, Luo H, Huang L, Luo H, Zhu X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell Int. 2020;20:16.
Silveira AB, Bidard FC, Kasperek A, Melaabi S, Tanguy ML, Rodrigues M, et al. High-accuracy determination of microsatellite instability compatible with liquid biopsies. Clin Chem. 2020;66(4):606–13.
Cortes-Ciriano I, Lee S, Park WY, Kim TM, Park PJ. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun. 2017;8:15180.
Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7(3):746–56.
Sun B. Current microsatellite instability testing in Management of Colorectal Cancer. Clin Colorectal Cancer 2020.
Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, et al. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the Association for Molecular Pathology and College of American pathologists. J Mol Diagn. 2017;19(3):341–65.
Luh F, Yen Y. FDA guidance for next generation sequencing-based testing: balancing regulation and innovation in precision medicine. NPJ Genom Med. 2018;3:28.
Georgiadis A, Durham JN, Keefer LA, Bartlett BR, Zielonka M, Murphy D, et al. Noninvasive detection of microsatellite instability and high tumor mutation burden in Cancer patients treated with PD-1 blockade. Clin Cancer Res. 2019;25(23):7024–34.
Gowen K, Clark TA, Gregg JP, Greene MZ, Murphy A, White J, et al. MSI-H testing via hybrid capture based NGS sequencing of liquid biopsy samples. J Clin Oncol. 2019;37(4_suppl):504.
Willis J, Lefterova MI, Artyomenko A, Kasi PM, Nakamura Y, Mody K, et al. Validation of microsatellite instability detection using a comprehensive plasma-based genotyping panel. Clin Cancer Res. 2019;25(23):7035–45.
Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(9):569.
Castle JC, Uduman M, Pabla S, Stein RB, Buell JS. Mutation-derived Neoantigens for Cancer immunotherapy. Front Immunol. 2019;10:1856.
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–46.
Riaz N, Morris L, Havel JJ, Makarov V, Desrichard A, Chan TA. The role of neoantigens in response to immune checkpoint blockade. Int Immunol. 2016;28(8):411–9.
McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–9.
Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–6.
Hartmaier RJ, Charo J, Fabrizio D, Goldberg ME, Albacker LA, Pao W, et al. Genomic analysis of 63,220 tumors reveals insights into tumor uniqueness and targeted cancer immunotherapy strategies. Genome Med. 2017;9(1):16.
Goodman AM, Castro A, Pyke RM, Okamura R, Kato S, Riviere P, et al. MHC-I genotype and tumor mutational burden predict response to immunotherapy. Genome Med. 2020;12(1):45.
Shim JH, Kim HS, Cha H, Kim S, Kim TM, Anagnostou V, et al. HLA-corrected tumor mutation burden and homologous recombination deficiency for the prediction of response to PD-(L)1 blockade in advanced non-small-cell lung cancer patients. Ann Oncol. 2020;31(7):902–11.
Choucair K, Morand S, Stanbery L, Edelman G, Dworkin L, Nemunaitis J. TMB: a promising immune-response biomarker, and potential spearhead in advancing targeted therapy trials. Review Cancer Gene Ther. 2020. https://doi.org/10.1038/s41417-020-0174-y
Fancello L, Gandini S, Pelicci PG, Mazzarella L. Tumor mutational burden quantification from targeted gene panels: major advancements and challenges. J Immunother Cancer. 2019;7(1):183.
Klempner SJ, Fabrizio D, Bane S, Reinhart M, Peoples T, Ali SM, et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist. 2020;25(1):e147–e59.
Merino DM, McShane LM, Fabrizio D, Funari V, Chen SJ, White JR, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer. 2020 Mar;8(1):e000147. https://doi.org/10.1136/jitc-2019-000147
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34.
Buttner R, Longshore JW, Lopez-Rios F, Merkelbach-Bruse S, Normanno N, Rouleau E, et al. Implementing TMB measurement in clinical practice: considerations on assay requirements. ESMO Open. 2019;4(1):e000442.
Merino DM, McShane L, Butler M, Funari VA, Hellmann MD, Chaudhary R, et al. TMB standardization by alignment to reference standards: Phase II of the Friends of Cancer Research TMB Harmonization Project. J Clin Oncol. 2019;37(15_suppl):2624.
Stenzinger A, Endris V, Budczies J, Merkelbach-Bruse S, Kazdal D, Dietmaier W, et al. Harmonization and standardization of panel-based tumor mutational burden measurement: real-world results and recommendations of the quality in pathology study. J Thorac Oncol. 2020;15(7):1177–89.
Vokes NI, Liu D, Ricciuti B, Jimenez-Aguilar E, Rizvi H, Dietlein F, et al. Harmonization of tumor mutational burden quantification and association with response to immune checkpoint blockade in non-small-cell lung Cancer. JCO Precis Oncol. 2019;3.
Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight. 2019;4(6).
Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133–50.
Keenan TE, Burke KP, Van Allen EM. Genomic correlates of response to immune checkpoint blockade. Nat Med. 2019;25(3):389–402.
Schrock AB, Ouyang C, Sandhu J, Sokol E, Jin D, Ross JS, et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann Oncol. 2019;30(7):1096–103.
Fabrizio DA, George TJ Jr, Dunne RF, Frampton G, Sun J, Gowen K, et al. Beyond microsatellite testing: assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J Gastrointest Oncol. 2018;9(4):610–7.
Bhangoo MS, Boasberg P, Mehta P, Elvin JA, Ali SM, Wu W, et al. Tumor mutational burden guides therapy in a treatment refractory POLE-mutant uterine Carcinosarcoma. Oncologist. 2018;23(5):518–23.
Johanns TM, Miller CA, Dorward IG, Tsien C, Chang E, Perry A, et al. Immunogenomics of Hypermutated Glioblastoma: a patient with Germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov. 2016;6(11):1230–6.
Lee L, Ali S, Genega E, Reed D, Sokol E, Mathew P. Aggressive-variant microsatellite-stable POLE mutant prostate Cancer with high mutation burden and durable response to immune checkpoint inhibitor therapy. JCO Precis Oncol. 2018;2:1–8.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–11.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–608.
Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377(25):2500–1.
Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus Ipilimumab in lung Cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093–104.
Singal G, Miller PG, Agarwala V, Li G, Kaushik G, Backenroth D, et al. Association of Patient Characteristics and Tumor Genomics with Clinical Outcomes among Patients with non-Small Cell Lung Cancer Using a Clinicogenomic database. JAMA. 2019;321(14):1391–9.
Paz-Ares L, Langer CJ, Novello S, Halmos B, Cheng Y, Gadgeel SM, et al. Pembrolizumab (pembro) plus platinum-based chemotherapy (chemo) for metastatic NSCLC: tissue TMB (tTMB) and outcomes in KEYNOTE-021, 189, and 407. Ann Oncol. 2019;30:v917–v8.
Marabelle A, Fakih MG, Lopez J, Shah M, Shapira-Frommer R, Nakgawa K, et al. Association of tumor mutational burden with outcomes in patients with select advanced solid tumors treated with PEMBROLIZUMAB in KEYNOTE-158. Ann Oncol. 2019;30:v477–8.
De Rubis G, Rajeev Krishnan S, Bebawy M. Liquid biopsies in Cancer diagnosis, monitoring, and prognosis. Trends Pharmacol Sci. 2019;40(3):172–86.
Johansson G, Andersson D, Filges S, Li J, Muth A, Godfrey TE, et al. Considerations and quality controls when analyzing cell-free tumor DNA. Biomol Detect Quantif. 2019;17:100078.
Merker JD, Oxnard GR, Compton C, Diehn M, Hurley P, Lazar AJ, et al. Circulating tumor DNA analysis in patients with Cancer: American Society of Clinical Oncology and College of American Pathologists Joint Review. J Clin Oncol. 2018;36(16):1631–41.
Nikolaev S, Lemmens L, Koessler T, Blouin JL, Nouspikel T. Circulating tumoral DNA: Preanalytical validation and quality control in a diagnostic laboratory. Anal Biochem. 2018;542:34–9.
Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24(9):1441–8.
Socinski M, Velcheti V, Mekhail T, Chae YK, Leal TA, Dowell JE, et al. LBA83 - final efficacy results from B-F1RST, a prospective phase II trial evaluating blood-based tumour mutational burden (bTMB) as a predictive biomarker for atezolizumab (atezo) in 1L non-small cell lung cancer (NSCLC). Ann Oncol. 2019;30:v919–v20.
Rizvi NA, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn MJ, et al. Durvalumab with or without Tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung Cancer: the MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 2020;6:661.
Li W, Wildsmith S, Ye J, Si H, Morsli N, He P, et al., editors. Plasma-based tumor mutational burden (bTMB) as predictor for survival in phase III EAGLE study: Durvalumab (D) ± tremelimumab (T) versus chemotherapy (CT) in recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC) after platinum failure. ASCO Virtual Scientific Program; 2020: American Society of Clinical Oncology.
U.S. Food and Drug Administration, FoundationOne® Liquid CDx 2020. Available from: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190032C.pdf. Accessed 10 May 2020.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50.
Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218.
Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, et al. Integrative analyses of colorectal Cancer show Immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44(3):698–711.
Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128–39.
Angell HK, Bruni D, Barrett JC, Herbst R, Galon J. The Immunoscore: Colon Cancer and beyond. Clin Cancer Res. 2020;26(2):332–9.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–40.
Auslander N, Zhang G, Lee JS, Frederick DT, Miao B, Moll T, et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med. 2018;24(10):1545–9.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–8.
Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, et al. A Cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175(4):984–97 e24.
Lu S, Stein JE, Rimm DL, Wang DW, Bell JM, Johnson DB, et al. Comparison of biomarker modalities for predicting response to PD-1/PD-L1 checkpoint blockade: a systematic review and meta-analysis. JAMA Oncol. 2019;5:1195.
Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362(6411).
Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359(6377):801–6.
Anagnostou V, Niknafs N, Marrone K, Bruhm DC, White JR, Naidoo J, et al. Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat Can. 2020;1(1):99–111.
Riaz N, Havel JJ, Kendall SM, Makarov V, Walsh LA, Desrichard A, et al. Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti-CTLA4 immunotherapy. Nat Genet. 2016;48(11):1327–9.
Fang W, Ma Y, Yin JC, Hong S, Zhou H, Wang A, et al. Comprehensive genomic profiling identifies novel genetic predictors of response to anti-PD-(L)1 therapies in non-small cell lung Cancer. Clin Cancer Res. 2019;25(16):5015–26.
Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung Cancer. Cancer Discov. 2017;7(12):1420–35.
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61.
Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8(1):1136.
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29.
McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung Cancer evolution. Cell. 2017;171(6):1259–71 e11.
Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359(6375):582–7.
Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397–404 e9.
Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7(2):188–201.
Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun. 2017;8:15440.
Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and Proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999–1008.
Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822–35.
Papillon-Cavanagh S, Doshi P, Dobrin R, Szustakowski J, Walsh AM. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open. 2020 Apr;5(2):e000706. https://doi.org/10.1136/esmoopen-2020-000706
George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine Leiomyosarcoma. Immunity. 2017;46(2):197–204.
Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–16.
Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5.
Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung Cancer: a retrospective analysis. Clin Cancer Res. 2016;22(18):4585–93.
Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung Cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36(7):633–41.
Dudnik E, Peled N, Nechushtan H, Wollner M, Onn A, Agbarya A, et al. BRAF mutant lung Cancer: programmed death ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check-point inhibitors. J Thorac Oncol. 2018;13(8):1128–37.
Braun DA, Ishii Y, Walsh AM, Van Allen EM, Wu CJ, Shukla SA, et al. Clinical validation of PBRM1 alterations as a marker of immune checkpoint inhibitor response in renal cell carcinoma. JAMA Oncol. 2019;5:1631.
Dizman N, Bergerot PG, Bergerot CD, Hsu J, Pal SK. Duration of treatment (DOT) with targeted therapies (TT) or immunotherapy (IO) in PBRM1 mutated metastatic renal cell carcinoma (mRCC). J Clin Oncol. 2019;37(7_suppl):622.
Hakimi AA, Ged Y, Flynn J, Hoen DR, Di Natale RG, Blum KA, et al. The impact of PBRM1 mutations on overall survival in greater than 2,100 patients treated with immune checkpoint blockade (ICB). J Clin Oncol. 2019;37(7_suppl):666.
Liu XD, Kong W, Peterson CB, McGrail DJ, Hoang A, Zhang X, et al. PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nat Commun. 2020;11(1):2135.
Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24(5):556–62.
Adashek JJ, Subbiah IM, Matos I, Garralda E, Menta AK, Ganeshan DM, et al. Hyperprogression and immunotherapy: fact, fiction, or alternative fact? Trends Cancer. 2020;6(3):181–91.
Matos I, Martin-Liberal J, Hierro C, Ochoa De Olza M, Viaplana C, Costa M, et al. Incidence and clinical implications of a new definition of hyperprogression (HPD) with immune checkpoint inhibitors (ICIs) in patients treated in phase 1 (Ph1) trials. J Clin Oncol. 2018;36(15_suppl):3032.
Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23(15):4242–50.
Singavi AK, Menon S, Kilari D, Alqwasmi A, Ritch PS, Thomas JP, et al. Predictive biomarkers for hyper-progression (HP) in response to immune checkpoint inhibitors (ICI); analysis of somatic alterations (SAs). Ann Oncol. 2017;28:v405.
Acknowledgments
We want to thank Bethany Sawchyn, Rachel Cunningham, and Jeffrey Venstrom for providing feedback on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editors: Baolin Zhang and Mario L. Rocci
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gjoerup, O., Brown, C.A., Ross, J.S. et al. Identification and Utilization of Biomarkers to Predict Response to Immune Checkpoint Inhibitors. AAPS J 22, 132 (2020). https://doi.org/10.1208/s12248-020-00514-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-020-00514-4