
Abstract
This article provides a theoretical case-study risk assessment report for a low-risk monoclonal antibody (mAb) therapeutic. In terms of risk, there are considerations around risks to safety, but also risks regarding effects on pharmacokinetics (PK), pharmacodynamics (PD), and efficacy. Much of the discussion in this document is around the risk of immunogenicity incidence. A higher incidence of immunogenicity would necessitate a detailed review of the PK, efficacy and safety in anti-drug antibody (ADA) positive and ADA negative subjects, in order to evaluate potential effects. The publication is intended to provide a framework of some the current thought processes around assessing immunogenicity risk and for building strategies to mitigate those risks. For this example, we have created a hypothetical antibody, ABC-123, targeting a membrane protein on antigen presenting cells, for the treatment of rheumatoid arthritis (RA). This hypothetical antibody therapeutic is provided as an example for the purposes of risk assessment for a low risk molecule, although any application of similar approach would be case by case.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A number of factors influence immunogenicity risk including: amino acid sequence, manufacturing processes, route and frequency of administration, disease indication, co-medications, and the mechanism of action (1,2). There is a general perception that humanized and fully human mAb therapeutics are considered low-risk for immunogenicity incidence. Humanized and fully human mAbs have a large amount of native sequence homology, and generally a low incidence of ADA in certain populations. A systematic review of published literature on marketed biologics for the treatment of chronic inflammatory diseases through November 2016 was conducted and reported by Strand et al., 2017 (3). Overall, 11 biologics were covered in the review, of which 8 were mAbs including: adalimumab, CT-P13 (an infliximab biosimilar), golimumab, infliximab, rituximab, secukinumab, tocilizumab, and ustekinumab. The antibody therapeutics discussed in the review included chimeric mouse-human antibodies (infliximab, rituximab), humanized antibodies (tocilizumab), and fully human antibodies (adalimumab, golimumab, secukinumab, and ustekinumab). The incidence of ADA formation in RA for the different classes of antibodies ranged significantly. It is understood that the incidence of ADA formation between biologics is not directly comparable, as the assays are unique, and the incidence can vary depending on a multitude of factors (4). Reported immunogenicity incidences are highly dependent on assay formats and related factors, such as sensitivity, drug tolerance, and interferences, such as the contribution from rheumatoid factor (5); however, this is an interest in following overall trends. In RA, the range of ADA detected for chimeric antibodies has been reported to be 0–62%, for humanized antibodies 0–16%, and for human antibodies 0–51%.
It is clear from these numbers that factors beyond the continued improvement in antibody engineering (the progression of chimeric to fully human antibodies) impact the incidence. Several reviews of available literature have shown that designing a fully human mAb, has not eliminated the immunogenicity concern (4,6,7). Even fully human antibody therapeutics with the same drug target, used to treat the same disease, can result in different rates of immunogenicity. Golimumab and adalimumab both target TNF-alpha and can be used for the treatment of RA. Surprisingly, they have different immunogenicity incidences in RA; golimumab (2–10%) and adalimumab (0–51%). A fully human antibody may still have the potential for immunogenicity, but what are some of the implications associated with immunogenicity?
There are several different types of risk associated with immunogenicity, primarily those related to efficacy and safety. For the drugs adalimumab, golimumab, infliximab rituximab, ustekinumab, and CT-P13, ADA positive patients displayed lower drug concentrations than ADA negative patients (3). RA patients who had binding ADA toward adalimumab, golimumab, infliximab, rituximab, or CT-P13 showed less improvement in disease activity (3). Generally speaking, some patients with ADA may have diminished clinical efficacy, often correlated to magnitude of ADA response or to the presence of neutralizing antibodies.
The safety risk from immunogenicity toward a fully human or humanized mAb is considered low, as the sequence is derived from endogenous human IgGs. IgG is present at high concentrations in serum, hence, the immune system is generally tolerized to IgG. However, ADA against some therapeutics in this class have demonstrated an increased potential for infusion-related reactions (3,8) and patients with ADA may be at higher risk for adverse events depending on the degree of sequence similarity, quality attributes, and mechanism of action. In spite of the potential immunogenicity-related outcomes discussed above, the risk-benefit has been considered sufficient to warrant approval of these biotherapeutics.
Hypothetical Case Study
Background
ABC-123 is a hypothetical fully human mAb IgG4 which acts as an immunosuppressant by antagonistically binding to cell surface receptors on dendritic cells (DC) and B-cells, for the treatment of RA with possible expansion into systemic lupus erythematosus (SLE) during development. The binding of ABC-123 to receptors on antigen presenting cells (APCs) prevents a costimulatory signal from being transferred to a T cell, thus suppressing the immune system in the case of autoimmune dysregulation. The IgG4 isotype was selected for its low effector function with relatively low Fc gamma receptor binding affinity, and it does not elicit antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC). IgG4 isotypes are known to undergo Fab arm exchange and as such ABC-123 was engineered with a S228P mutation to abrogate Fab arm exchange. ABC-123 will be manufactured in a mammalian (CHO) cell line. The expected dosing paradigm in RA includes twice-monthly subcutaneous injections (SC). Information in this submission is provided to support the first in human clinical trial for ABC-123 in healthy subjects.
Immunogenicity Risk
The risk assessment of ABC-123 immunogenicity was prepared in alignment with the FDA guidance on Immunogenicity Testing of Therapeutic Protein Products, Jan 2019 (9) and the EMA Guideline on Immunogenicity Assessment of Therapeutic Proteins, May 2017 (10).
Sequence (Predictive Tools) and Structural Based Risk
ABC-123 is a human IgG4 isotype mAb. An IgG4 was selected for its low effector function with relatively low Fc gamma receptor binding affinity, and it does not elicit ADCC or CDC. Ig4 isotypes are known to undergo Fab arm exchange and as such, ABC-123 was engineered with an S228P mutation in the Fc region to abrogate the Fab exchange (11). The hypothetical amino acid sequence of ABC-123 is provided as supplementary material 1.
An in-silico immunogenicity analysis of the sequence-based risk of ABC-123 was performed to enable selection of the drug candidate with the least immunogenicity risk. HLA-DR binding epitopes and clusters were identified in the light and heavy chains using the Epimatrix algorithm (12). All of the epitopes identified were epitopes that were also present in multiple germline IgG4 sequences. As there were no non-germline epitopes identified in the mature sequence of ABC-123 that were predicted to bind with high affinity to HLA-DR, hence, no in vitro immunogenicity assays were performed. Although in vitro immune cell based assays were not performed in this case study, they can assist in the identification of therapeutic proteins which activate T cells (13) and thereby contribute to the risk assessment for immunogenicity incidence.
The S228P mutation is non-self, however, the Epimatrix algorithm did not predict binding of this residue to HLA-DR alleles, in the context of the 8 flanking residues either preceding or following the mutation. Several marketed IgG4 mAbs such as nivolumab, pembrolizumab, and inotuzumab ozogamicin have this mutation (14,15) and also have relatively low immunogenicity incidences 11.2%, 2.1%, 3% (16,17,18). In sum, the mAb ABC-123 had no substantial sequence-based regions of concern that are expected to influence immunogenicity incidence.
The sequence-based risk provides insight into the immunogenic potential of the molecule and when coupled with the lower immunogenicity observed in the clinical experience of other IgG4 mAbs containing the S228P mutation, it suggests minimal incidence risk. However, the impact that an immune response can have on exposure and efficacy is a separate risk that is currently difficult to estimate. The clinical relevance of an immune response will be evaluated as the program progresses, by assessing the impact of ADA on PK, PD, efficacy, and safety.
Product Quality Attributes Based Risk
ABC-123 was manufactured using a Good Manufacturing Practices (GMP) CHO-based cell line. For clinical studies, ABC-123 was manufactured according to FDA CMC and GMP manufacturing guidelines and met all the requirements for critical product quality attributes as outlined in the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q6B specification document (19). All product structural variants and process related impurities were within specifications and in line with previously administered mAbs manufactured by the sponsor. No formulation-based immunogenicity risks are anticipated for ABC-123. Beyond factors previously published, there are no novel critical quality attributes likely to affect immunogenicity incidence.
Mechanism of Action-Based Risk
B and T cell interactions are important for immune system regulation. The pharmacological target of ABC-123, is expressed on APCs (Fig. 1), which may lead to an increased risk of immunogenicity due to the fact that APCs uptake foreign proteins and present them as antigens. However, since the ABC-123 is an antagonist, it should decrease the immune response by blocking the signal generated by interactions with T cells, as long as it is present at levels above a therapeutic threshold. The immune suppression mechanism of action is likely to favor a low immunogenicity incidence.

Mechanism of Action of ABC-123
Population Based Risk
While ABC-123 is expected to have minimal immunogenicity in healthy subjects, the likelihood of an immune response may be higher in an autoimmune population such as RA due to heightened immune activity. As ABC-123 reaches therapeutic levels for a sustained period of time and down, regulates the immune system in the multiple dose trial in patients with RA, a reduction in immunogenicity may be observed compared to initial treatment.
Conditions of Use Based Risk
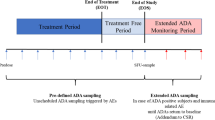
ABC-123 is predicted to have a half-life of ~14 days in humans. The planned dosing paradigm for the first in human study 01 will be a rising single dose in healthy subjects with intravenous (IV) doses of 0.1, 0.3, 1, 3, and 10 mg/kg and SC doses of 1 and 3 mg/kg. PK and ADA will be monitored for 4 months. Sampling for ADA will occur at baseline and post dose on Days 14, 28, 56, 84, and 112. PK samples will be collected at all ADA sampling time points. A simulation of the ABC-123 concentration following a 10 mg/kg single IV dose is shown in Fig. 2a.

Simulations of ABC-123 Concentrations Following Administration of the Highest Expected Single Dose (10 mg/kg) (a) or Repeated Dose (200 mg) Every 14 Days for 3 Months (b)
Following study 01, will be a study 02 in RA patients with SC dosing every 2 weeks for 3 months with doses of 25 mg, 100 mg, 200 mg (dosing levels may change based on safety outcomes from study 01). In this study ADA sample collection will occur at baseline, and pre-dose on days 14, 28, 56, and 84 with a sample taken at 3 months (Day 168) following the last dose. PK samples will be collected at all ADA sampling time points. Sampling at 3 months following the last dose is to ensure that the ADA samples are collected at low levels of ABC-123. A simulation of the ABC-123 concentration following repeated doses of 200 mg SC is shown in Fig. 2b. It is possible that the repeated dosing may leads to increased incidence of ADA compared to that following a single dose; however, multiple dosing is a necessity for chronic diseases such as RA. The dosing and sampling paradigm for ABC-123 is representative of other clinical programs in RA and is not expected to have a meaningful effect on immunogenicity incidence. Interestingly, it has also been shown that intermittent dosing can lead to higher incidences of immunogenicity (20,21,22).
The standard of care for RA includes treatment with immune suppressants such as methotrexate. Studies in RA patients with several anti-TNF antibodies such as infliximab, adalimumab, and golimumab have shown that the methotrexate reduced immunogenicity incidence (23,24,25,26). Hence the observed immunogenicity in this target population which is likely to be co-dosed with an immunosuppressant, may in fact, be lower than what would be observed without the comedication.
Immunogenicity Strategy
Overall, the anticipated immunogenicity risk, both prevalence (frequency at baseline) and incidence, is considered low for ABC-123. As such, clinical sampling will reflect a low immunogenicity risk situation and dose escalation will proceed without the need to review immunogenicity results following the previous dose of ABC-123. Since immunogenicity risk is low, initial clinical studies will be carried out in healthy volunteers before transitioning to the RA population. Immunogenicity incidence will be compared across dose levels and between routes of administration (IV and SC). The effect of ADA on PK, PD, efficacy, and safety will be assessed.
Timing of NAb Assay Development and Justification
In early phase studies, PD markers (receptor occupancy of the drug target) will be used to monitor for the presence of neutralizing antibodies (NAb). In subjects who are positive for NAb, the receptor occupancy is expected to decrease, as compared to that in negative subjects. If the receptor occupancy assay proves to be sensitive and robust, data will be presented at the End of Phase 2 meeting to propose using it in place of a NAb assay (9). If the receptor occupancy assay is not considered sensitive enough, a competitive ligand binding-based NAb assay will be proposed to support Phase 3 studies, in accordance with industry guidelines for antagonistic biotherapeutics (27) at the End of Phase 2 discussions with health authorities.
Immunogenicity Assays
Serum samples will be evaluated for the presence of ADA through use of bridging immunoassays. Immunogenicity assays will be validated in accordance with FDA and EMA guidance and industry standards. The first tier of the immunogenicity testing is a screening assay to detect ADA to ABC-123. Samples testing positive in the screening assay will be subsequently tested in a confirmatory assay (second tier), by competition with excess ABC-123, to demonstrate that the ADA response is specific for the therapeutic protein product. Samples that are confirmed positive will be diluted further to obtain a value in titer units that is defined as log10 (dilution factor). The screening and confirmatory assays will target false positive rates of 5% and 1%, respectively (9). The targeted drug tolerance level of the screening assay will be such that 100 ng/mL of ADA positive control (9) can be detected in presence of at least 50 μg/mL of ABC-123, which is above the expected range of steady state trough drug concentrations for a 200 mg repeated SC dose, Fig. 2b. Previous assessments have shown that there is no soluble form of the target receptor detectable in circulation in humans and as such, interference of soluble drug target in the immunogenicity assay is not expected.
ADA Assay
Description
Validated ADA assays will be developed and run in alignment with the FDA (2019) and EMA (mAb) guidance for immunogenicity with respect to assay performance characteristics, including sensitivity, specificity, and drug tolerance at relevant clinical levels (9,10).
Briefly, ADA against ABC-123 will be detected by a bridging ELISA or electrochemiluminescence assay, as the bridging format is expected to yield a sufficient level of tolerance to drug. An affinity-purified anti-idiotypic polyclonal rabbit antibody directed against the complementarity determining regions of ABC-123 will serve as positive control reagent. A bridging assay typically involves the labeling of the drug by two different molecular tags, one being typically a biotin, which is used for capture, and another used for detection; however, other detection schemes are also commonly used. When an ADA response bridges both drug moieties, the assay is able to detect a complex. Several different ADA screening assay formats are described in the literature (28,29,30,31,32).
For confirmatory assays, samples will be pre-incubated overnight with excess drug, prior to incubation in assay diluent containing both biotinylated and reporter conjugated ABC-123. All subsequent steps will be similar to the screening assay procedure described above.
Screening and the confirmatory cut-points will be determined during validation by analyzing 50 individual drugs naive serum samples (in the absence and the presence of spiked ABC-123). The screening and the confirmatory cut-points will be determined nonparametrically according to Devanarayan et al, 2017 and Shankar et al., 2008 (33,34) or other appropriate statistical models, based on the data.
As the disease indication of RA can have interference from rheumatoid factor, additional assay design and performance considerations will be evaluated, such as the use of rheumatoid factor blocking reagents (5). ADA assay cut-points will be determined for the healthy population and will be updated for the RA population, as interference from disease characteristics is possible.
Timing of ADA Assay Result Turnaround Times
As ABC-123 represents a low immunogenicity risk, dose escalation will proceed without analysis of immunogenicity results. Immunogenicity data will be analyzed at an interim time and at the end of the study for all Phase 1 studies.
NAb Assay
In early development (Phase 1 and Phase 2), a receptor occupancy assay will be used to monitor for potential effects of NAb development, although the receptor occupancy assay may not be appropriate for a Phase 3 setting. Other biomarkers related to clinical efficacy will be studied during early clinical development. If a robust efficacy biomarker is identified, the need for a NAb assay may be minimal. A plate-based competitive ligand binding NAb assay will be developed as a back-up. Due to the antagonistic nature of ABC-123, and its low immunogenicity risk, a cell-based NAb assay will not be developed. The NAb assay validation will follow the recommendations in the regulatory guidance and will include the determination of a cut-point (1% false positive rate), sensitivity, selectivity, and drug tolerance. This assay will be developed prior to Phase 3 and the NAb strategy will be discussed with health authorities at the end of Phase 2 meetings.
Immunogenicity Results
Preclinical Results
A 3 months toxicology study was conducted in cynomolgus monkeys. ABC-123 has similar affinity to its target receptors in both humans and cynomolgus monkeys, making monkey a relevant species for toxicology assessment. In the 3 months dosing period, 9 of 36 (25%) monkeys developed ADA and no ADA related safety events were observed. As ABC-123 is a fully human mAb, immunogenicity was expected in monkeys but is not predictive of clinical immunogenicity (35).
Clinical Results
No clinical data are available.
Conclusions of Risk Assessment for ABC-123
In summary, ABC-123 is a fully human mAb with no substantial sequence-based regions of concern that is expected to influence immunogenicity incidence. There are no Critical Quality Attributes that have been identified with ABC-123 that might result in an increased risk of immunogenicity. The immune suppression mechanism of action is likely to favor a low incidence, although the likelihood of an immune response may be higher in an autoimmune population such as RA due to heightened immune activity. As standard of care for some RA patients may also involve treatment with immunosuppressants, it is difficult to estimate the immunogenicity incidence in this target population. The dosing and sampling paradigm for ABC-123 is representative of other clinical programs in RA and is not expected to have a meaningful effect on immunogenicity incidence. Thus, the overall risk of immunogenicity for ABC-123 is low.
Discussion
General Recommendations for mAbs
Limited Structural Modifications
Initial clinical studies with human/mouse chimeric mAbs such as infliximab and rituximab showed a moderate to high clinical immunogenicity incidence (20–51% in psoriasis and 11% in RA, respectively), suggesting that the more human-like the sequence, the lower the immunogenicity (36,37). Most humanized and fully human mAbs have a lower incidence of ADA than do chimeric mAbs (38), although having a fully human amino acid sequence does not ensure a lack of immunogenicity (4,7). Individual amino acid changes such as S228P to reduce Fab exchange in IgG4 for nivolumab, pembrolizumab, and inotuzumab ozogamicin showed relatively low incidences of ADA 11.2%, 2.1%, 3% (16,17,18). Likewise, amino acid substitutions to increase the half-life of mAbs (motavizumab-YTE, MEDI8897, VRC01LS) have also not have not led to high immunogenicity incidences, 25%, 13.7%, and 0%, respectively (39,40,41). It is worth noting that the ADA data for these three molecules with half-life extension modifications were reported based on data from Phase 1 trials, hence the number of subjects was not large. An additional example is eculizumab which was engineered as a hybrid Fc IgG2/4 to reduce effector function and complement activation (42), which showed an immunogenicity incidence of 2% (43). ABC-123 is a fully human mAb with no substantial sequence-based regions of concern that are expected to influence immunogenicity incidence.
Quality Attributes
Unlike small molecule therapeutics, biologics present some unique challenges and features. Host cells are genetically manipulated to produce the therapeutic of interest. When these products are recombinant antibodies, they typically require purification from other cell culture components, prior to formulation. Therefore, immunogenicity, which is an unwanted immune response to the therapeutic, may result from product related variants (44), or from raw materials or process related impurities which co-purify with the therapeutic drug molecule of interest (45,46).
It is important to identify critical quality attributes that may impact immunogenicity, early during the development process and establish Quality by Design procedures to proactively address and establish manufacturing controls (47). ICH guidance documents Q8–11 address these points (48). Process-related impurities may have a serious impact on immunogenicity, particularly for repeated dose studies. Of particular interest are raw materials, product variants, host cell proteins, host cell DNA, and possible leached materials from the purification process. Establishing specifications for the measured levels of each factor that might impact immunogenicity is imperative, as part of the process controls during manufacture. In the case of ABC-123, there are no identified novel product quality attributes that are likely to affect immunogenicity incidence.
Mechanisms of Action
In the example of ABC-123, the mechanism of action is to reduce immune system function in RA patients. The immunogenicity observed in marketed products used to treat chronic inflammatory diseases has been reviewed; for humanized or fully human mAbs (with the exception of adalimumab), the immunogenicity incidences have been less than 15% (3).
Several reviews have summarized the incidence of immunogenicity in oncology products (49,50,51). Some of the mAbs used in oncology employ an immune stimulatory mechanism of action; hence the incidence of immunogenicity could theoretically be higher than for a mAb with a non-immune stimulatory mechanism. Monoclonal antibodies targeting check point inhibitors such as CTLA-4 or PD-1/PDL1 have been developed to induce anti-tumor immune responses in cancer patients. Thus far, the marketed immune check point inhibitor mAbs (anti CTLA-4: ipilimumab; anti-PD-1: nivolumab, pembrolizumab, cemiplimab; and anti-PDL-1: avelumab, durvalumab) showed relatively low immunogenicity incidences (0–12.7%) (50). The exception in this therapeutic area is atezolizumab which reported an incidence of 39.1% across multiple clinical studies (50). Atezolizumab is an IgG1, anti-PD-L1 inhibitor that has an engineered Fc domain (N298A) to prevent binding to Fcγ receptors (52). Some immune stimulatory mAbs in oncology have tested in combination, such as nivolumab and ipilimumab. The incidence of ADA to nivolumab as monotherapy was 11.2% and in combination therapy with ipilimumab was 26.0% to 37.8% (16), suggesting that the multiple immune stimulation mechanisms may increase ADA incidence, although more data are needed to know if this finding can be generalized.
Another mechanism of action used in the oncology therapeutic area is that of B cell depleting mAbs, such as rituximab, obinutuzumab or veltuzumab. The expectation would be that pharmacologically blocking the humoral response would reduce the overall ADA incidence to this class of drug. Multiple studies of therapeutics with this mechanism of action have been reviewed by van Brummelen et al, 2016, and they note that despite the mechanism of B cell depletion, ADA formation has been detected in some instances (49).
In addition to immunosuppressive and immune-stimulating mechanisms of action, there are also mAbs which have mechanisms of action that are not designed to affect the immune system. Examples of non-immune related mechanisms of action in approved drugs include anti-infectives such as palivizumab, bezlotoxumab, and ibalizumab with incidences of < 2, 0 and < 1%, respectively (53,54,55,56). Another set of examples includes mAbs that target PCSK9. In this mechanism of action (blocking PCSK9) a wide range of ADA incidence was observed for evolocumab, alirocumab, and bococizumab; 0.3, 4.8 and 48%, respectively (57,58,59). Presumably factors other than mechanism of action gave rise to the higher incidence for bococizumab, as the other two mAbs with the same mechanism showed substantially lower incidence.
Observations which Could Lead to a Re-evaluation of the Risk Strategy
Although the risk associated with a mAb to induce an immunogenicity response is generally classified as low, the immune repertoire of individuals within any disease indication will be highly relevant in determining the actual immunogenicity developed to a therapeutic molecule. While the ADA incidences can vary greatly due to specific assay conditions and are not directly comparable between molecules, if a high incidence rate is observed during a clinical study, it may prompt a more detailed exploration of immunogenicity assessment. Specifically, if ADA titers are high or increasing during a multiple dose study or are persistent in nature, these would prompt a re-evaluation of the immunogenic potential of the drug molecule.
The clinical impact of an ADA response may provide a compelling re-assessment of the immunogenicity risk. Specifically, if there is impact on drug exposure or the PD response, this may trigger more frequent sampling to determine the duration of the ADA response or further characterization of the ADA response to determine neutralization activity. The proportion of subjects who develop a loss in exposure as correlated to ADA positive status would add granularity to the course of action to be taken. While it is theoretically possible to change dose levels or dosing frequency, there may not be a large amount of flexibility, depending on the stage of the program, as efficacy, safety, and patient convenience must also be considered. Therapeutic drug monitoring is sometimes used for approved TNF inhibitor drugs that are on the market, to modify the dose when a subject loses response due to ADA. Although, a caveat to this approach is that the ADA and PK assay methods used for therapeutic monitoring are generally different from those used by the sponsor in clinical trials (60).
Infusion/injection site reactions or hypersensitivity reactions are common with antibody therapeutics. If these safety events are accompanied with pre-existing immunity to the therapeutic, there may be a reason to pre-screen individuals for titer to drug. If serious hypersensitivity adverse events, such as anaphylaxis, are observed, it may warrant developing an IgE assay to characterize the response (61). The observed frequency of immunogenicity related safety events in early trials and the potential risk/benefit of screening for pre-existing immunogenicity would be discussed with health authorities at an end of Phase 2 meeting. In the case of less serious clinical safety events, that appear to be related to the ADA response, it may more appropriate to determine whether the ADA response is persistent and follow patients until such time as their titer drops below the level of detection of the ADA assay, to see if the clinical adverse event persists after the ADA response has resolved.
Clinically impactful responses that can be linked to ADA development would provide the most compelling rationale for re-evaluation of the immunogenicity risk assessment. If adverse events are serious enough and correlated with ADA positive subjects, a re-assessment of immunogenicity risk would likely be triggered. In these situations, it may be important to modify the risk assessment, to pursue a more cautious course of action. The accompanying assay strategy should be modified accordingly. Depending on the severity of the safety signals observed, dosing may be halted, or the program may be terminated altogether. In the specific context of RA (which has several available treatment options), if serious immunogenicity related safety events were to be observed, this could cause termination of the program.
Future Perspectives
Although this document describes a rather simplified case study for a therapeutic biomolecule, as the field of biologics evolves, so will the risk assessment and mitigation strategies. As mAbs are developed for new pharmacological targets, uncertainties in the pharmacology of the target can also lead to uncertainties in the risk assessment of immunogenicity, particularly in the immune-oncology therapeutic area.
In addition, for well characterized therapeutic proteins such as mAbs, there is the possibility of using a sensitive PD assay that reflects clinical activity, in lieu of a NAb assay (9). One reason for assessing the NAb is to inform an immunogenicity-related rationale for unexpected reductions in clinical efficacy. If a therapeutic protein program has a sensitive PD assay with low variability that is highly correlated with clinical efficacy, the PD marker itself may be able to show the degree of NAb-related reductions in efficacy in ADA positive subjects.
Conclusions
In response to recent guidance from FDA in 2019 and EMA in 2017, a hypothetical case study of an immunogenicity risk assessment for a mAb drug candidate (ABC-123) has been prepared for illustrative purposes, as if it were to be submitted in support of a first in human clinical study. Given that ABC-123 is a mAb without sequence or quality issues that are likely to impact ADA formation and that ABC-123 has an immunosuppressive mechanism of action, the risk of immunogenicity incidence was considered low. Some caveats were presented for situations or cases in which the risk for a mAb could increase. The process of outlining immunogenicity concerns for new drug candidates may help to guide the development of the program and the overall risk to the first in human subjects.
References
Koren E, Smith HW, Shores E, Shankar G, Finco-Kent D, Rup B, et al. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J Immunol Methods. 2008;333(1–2):1–9.
Shankar G, Pendley C, Stein KE. A risk-based bioanalytical strategy for the assessment of antibody immune responses against biological drugs. Nat Biotechnol. 2007;25(5):555–61.
Strand V, Balsa A, Al-Saleh J, Barile-Fabris L, Horiuchi T, Takeuchi T, et al. Immunogenicity of biologics in chronic inflammatory diseases: a systematic review. BioDrugs. 2017;31(4):299–316.
Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9(10):767–74.
Araujo J, Zocher M, Wallace K, Peng K, Fischer SK. Increased rheumatoid factor interference observed during immunogenicity assessment of an fc-engineered therapeutic antibody. J Pharm Biomed Anal. 2011;55(5):1041–9.
Getts DR, Getts MT, McCarthy DP, Chastain EM, Miller SD. Have we overestimated the benefit of human(ized) antibodies? MAbs. 2010;2(6):682–94.
Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2(3):256–65.
Caceres MC, Guerrero-Martin J, Perez-Civantos D, Palomo-Lopez P, Delgado-Mingorance JI, Duran-Gomez N. The importance of early identification of infusion-related reactions to monoclonal antibodies. Ther Clin Risk Manag. 2019;15:965–77.
Immunogenicity Testing of Therapeutic Protein Products —Developing and Validating Assays for Anti-Drug Antibody Detection [Internet]. 2019. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/immunogenicity-testing-therapeutic-protein-products-developing-and-validating.
Guideline on Immunogenicity assessment of therapeutic proteins [Internet]. 2017. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf.
Silva JP, Vetterlein O, Jose J, Peters S, Kirby H. The S228P mutation prevents in vivo and in vitro IgG4 fab-arm exchange as demonstrated using a combination of novel quantitative immunoassays and physiological matrix preparation. J Biol Chem. 2015;290(9):5462–9.
Mufarrege EF, Giorgetti S, Etcheverrigaray M, Terry F, Martin W, De Groot AS. De-immunized and functional therapeutic (DeFT) versions of a long lasting recombinant alpha interferon for antiviral therapy. Clin Immunol. 2017;176:31–41.
Joubert MK, Deshpande M, Yang J, Reynolds H, Bryson C, Fogg M, et al. Use of in vitro assays to assess immunogenicity risk of antibody-based biotherapeutics. PLoS One. 2016;11(8):e0159328.
Zhang T, Song X, Xu L, Ma J, Zhang Y, Gong W, et al. The binding of an anti-PD-1 antibody to FcgammaRI has a profound impact on its biological functions. Cancer Immunol Immunother. 2018;67(7):1079–90.
Herbener P, Schonfeld K, Konig M, Germer M, Przyborski JM, Bernoster K, et al. Functional relevance of in vivo half antibody exchange of an IgG4 therapeutic antibody-drug conjugate. PLoS One. 2018;13(4):e0195823.
OPDIVO (nivolumab) injection, for intravenous use [Internet]. 2018. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf.
KEYTRUDA (pembrolizumab) for injection, for intravenous use [Internet]. 2019. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125514Orig1s054lbl.pdf.
BESPONSA (inotuzumab ozogamicin) for injection, for intravenous use [Internet]. 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf.
Specifications: test procedures and acceptance criteria for biotechnological/biological products Q6B [Internet]. 1999. Available from: https://database.ich.org/sites/default/files/Q6B_Guideline.pdf.
Maser EA, Villela R, Silverberg MS, Greenberg GR. Association of trough serum infliximab to clinical outcome after scheduled maintenance treatment for Crohn's disease. Clin Gastroenterol Hepatol. 2006;4(10):1248–54.
Ordas I, Mould DR, Feagan BG, Sandborn WJ. Anti-TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics-based dosing paradigms. Clin Pharmacol Ther. 2012;91(4):635–46.
Schreiber S, Khaliq-Kareemi M, Lawrance IC, Thomsen OO, Hanauer SB, McColm J, et al. Maintenance therapy with certolizumab pegol for Crohn's disease. N Engl J Med. 2007;357(3):239–50.
Maini RN, Breedveld FC, Kalden JR, Smolen JS, Davis D, Macfarlane JD, et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998;41(9):1552–63.
Pouw MF, Krieckaert CL, Nurmohamed MT, van der Kleij D, Aarden L, Rispens T, et al. Key findings towards optimising adalimumab treatment: the concentration-effect curve. Ann Rheum Dis. 2015;74(3):513–8.
Zhuang Y, Xu Z, Frederick B, de Vries DE, Ford JA, Keen M, et al. Golimumab pharmacokinetics after repeated subcutaneous and intravenous administrations in patients with rheumatoid arthritis and the effect of concomitant methotrexate: an open-label, randomized study. Clin Ther. 2012;34(1):77–90.
Wang W, Leu J, Watson R, Xu Z, Zhou H. Investigation of the mechanism of therapeutic protein-drug interaction between methotrexate and Golimumab, an anti-TNFalpha monoclonal antibody. AAPS J. 2018;20(3):63.
Wu B, Chung S, Jiang XR, McNally J, Pedras-Vasconcelos J, Pillutla R, et al. Strategies to determine assay format for the assessment of neutralizing antibody responses to biotherapeutics. AAPS J. 2016;18(6):1335–50.
Peng K, Siradze K, Quarmby V, Fischer SK. Clinical immunogenicity specificity assessments: a platform evaluation. J Pharm Biomed Anal. 2011;54(3):629–35.
Carrasco-Triguero M, Dere RC, Milojic-Blair M, Saad OM, Nazzal D, Hong K, et al. Immunogenicity of antibody-drug conjugates: observations across eight molecules in eleven clinical trials. Bioanalysis. 2019.
Qiu ZJ, Ying Y, Fox M, Peng K, Lewin-Koh SC, Coleman D, et al. A novel homogeneous biotin-digoxigenin based assay for the detection of human anti-therapeutic antibodies in autoimmune serum. J Immunol Methods. 2010;362(1–2):101–11.
Gross J, Moller R, Henke W, Hoesel W. Detection of anti-EPO antibodies in human sera by a bridging ELISA is much more sensitive when coating biotinylated rhEPO to streptavidin rather than using direct coating of rhEPO. J Immunol Methods. 2006;313(1–2):176–82.
Mikulskis A, Yeung D, Subramanyam M, Amaravadi L. Solution ELISA as a platform of choice for development of robust, drug tolerant immunogenicity assays in support of drug development. J Immunol Methods. 2011;365(1–2):38–49.
Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–81.
Devanarayan V, Smith WC, Brunelle RL, Seger ME, Krug K, Bowsher RR. Recommendations for systematic statistical computation of immunogenicity cut points. AAPS J. 2017;19(5):1487–98.
FDA. S6 Addendum to Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals. 2012.
REMICADE (infliximab) Lyophilized Concentrate for Injection, for Intravenous Use [Internet]. 2013. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/103772s5359lbl.pdf.
RITUXAN (rituximab) Injection for Intravenous Use [Internet]. 2010. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/103705s5311lbl.pdf.
Roskos LK, Davis CG, Schwab GM. The clinical pharmacology of therapeutic monoclonal antibodies. Drug Dev Res. 2004;61(3):108–20.
Robbie GJ, Criste R, Dall'acqua WF, Jensen K, Patel NK, Losonsky GA, et al. A novel investigational fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. 2013;57(12):6147–53.
Griffin MP, Khan AA, Esser MT, Jensen K, Takas T, Kankam MK, et al. Safety, Tolerability, and Pharmacokinetics of MEDI8897, the Respiratory Syncytial Virus Prefusion F-Targeting Monoclonal Antibody with an Extended Half-Life, in Healthy Adults. Antimicrob Agents Chemother. 2017;61(3).
Gaudinski MR, Coates EE, Houser KV, Chen GL, Yamshchikov G, Saunders JG, et al. Safety and pharmacokinetics of the fc-modified HIV-1 human monoclonal antibody VRC01LS: a phase 1 open-label clinical trial in healthy adults. PLoS Med. 2018;15(1):e1002493.
Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25(11):1256–64.
SOLIRIS™ (eculizumab) Concentrated solution for intravenous infusion [Internet]. 2007. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/125166lbl.pdf.
Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. 2016;2016:1298473.
Vanderlaan M, Zhu-Shimoni J, Lin S, Gunawan F, Waerner T, Van Cott KE. Experience with host cell protein impurities in biopharmaceuticals. Biotechnol Prog. 2018;34(4):828–37.
Fischer SK, Cheu M, Peng K, Lowe J, Araujo J, Murray E, et al. Specific immune response to phospholipase B-like 2 protein, a host cell impurity in Lebrikizumab clinical material. AAPS J. 2017;19(1):254–63.
Alt N, Zhang TY, Motchnik P, Taticek R, Quarmby V, Schlothauer T, et al. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biologicals. 2016;44(5):291–305.
ICH Quality Implementation Working Group, Points To Consider (R2); ICH-Endorsed Guide ForICH Q8/Q9/Q10 Implementation [Internet]. 2011. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_9_10_QAs/PtC/Quality_IWG_PtCR2_6dec2011.pdf.
van Brummelen EM, Ros W, Wolbink G, Beijnen JH, Schellens JH. Antidrug antibody formation in oncology: clinical relevance and challenges. Oncologist. 2016;21(10):1260–8.
Davda J, Declerck P, Hu-Lieskovan S, Hickling TP, Jacobs IA, Chou J, et al. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J Immunother Cancer. 2019;7(1):105.
Talotta R, Rucci F, Canti G, Scaglione F. Pros and cons of the immunogenicity of monoclonal antibodies in cancer treatment: a lesson from autoimmune diseases. Immunotherapy. 2019;11(3):241–54.
Deng R, Bumbaca D, Pastuskovas CV, Boswell CA, West D, Cowan KJ, et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs. 2016;8(3):593–603.
SYNAGIS® (palivizumab) injection, for intramuscular use [Internet]. 2014. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/103770s5185lbl.pdf.
ZINPLAVA™ (bezlotoxumab) injection, for intravenous use [Internet]. 2016. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761046s000lbl.pdf.
Montgomery DL, Matthews RP, Yee KL, Tobias LM, Dorr MB, Wrishko RE. Assessment of Bezlotoxumab Immunogenicity. Clin Pharmacol Drug Dev. 2019.
Markham A. Ibalizumab: First Global Approval. Drugs. 2018;78(7):781–5.
REPATHA (evolocumab) injection, for subcutaneous use [Internet]. 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125522s014lbl.pdf.
PRALUENT (alirocumab) injection, for subcutaneous use [Internet]. 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125559Orig1s000lbledt.pdf.
Ridker PM, Tardif JC, Amarenco P, Duggan W, Glynn RJ, Jukema JW, et al. Lipid-reduction variability and antidrug-antibody formation with bococizumab. N Engl J Med. 2017;376(16):1517–26.
Kopylov U, Ben-Horin S, Seidman E. Therapeutic drug monitoring in inflammatory bowel disease. Ann Gastroenterol. 2014;27(4):304–12.
Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358(11):1109–17.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editor: Johanna Mora
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 24 kb)
Rights and permissions
About this article

Cite this article
Kernstock, R., Sperinde, G., Finco, D. et al. Clinical Immunogenicity Risk Assessment Strategy for a Low Risk Monoclonal Antibody. AAPS J 22, 60 (2020). https://doi.org/10.1208/s12248-020-00440-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-020-00440-5