
Abstract
Background
A quality by design approach can potentially lead to a more robust/rugged method development due to emphasis on the risk assessment and management. By carefully understanding the step-by-step procedure for analytical QbD-based optimization parameters, such as analytical target profile and critical quality attributes (CQAs), was assessed. The present study describes the simple, rapid, sensitive and cost-effective RP-HPLC method development and validation for the estimation of evogliptin tartrate in pharmaceutical dosage form.
Results
The factor screening studies were performed using Box–Behnken design by three key components of the RP-HPLC method (mobile phase, pH and flow rate). The chromatographic conditions were optimized with the Design Expert software trial version 13.0. The optimal chromatographic separation was achieved having water C18 column (250 mm × 4.6 mm, 5 μ) and using mobile phase as a methanol and phosphate buffer (pH 4.5) 60:40% v/v with a flow rate 1.0 ml/min and UV detection at 267 nm. The Box–Behnken experimental design describes the interrelationship of mobile phase, pH and flow rate at three different levels, and responses of retention time and tailing factor were observed with response surface plot and statistical data. The developed method was validated as per recommended ICH guidelines which revealed the high degree of linear, precise, accurate, sensitive and robust method over the existing RP-HPLC method for evogliptin tartrate.
Conclusion
The developed QbD-based method helped in generating a design space and operating space with knowledge of all method performance characteristics, and RP-HPLC method takes less time and can be used in the industry for routine quality control of bulk and marketed formulation of evogliptin tartrate.
Similar content being viewed by others

Background
Evogliptin tartrate is a potent orally bioavailable and selective class of DPP-IV (dipeptidyl phosphate-IV) inhibitors with anti-diabetic class. In clinical practice, gliptins have proved to be safe and effective oral drugs reducing glucose level in type-2 diabetes patients [1–3]. EVO is a DPP-IV inhibitors that reduce degradation of endogenous glucagon-like peptide 1 (GLP-1) to increase insulin secretion and decrease glucagon. Evogliptin tartrate is effective in improving glycosylated haemoglobin (HbA1c) and fasting plasma glucose without inducing hypoglycaemia events [4–7].
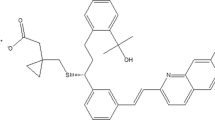
IUPAC name of EVO is (3R)-4-[(3R)-3-amino-4-(2,4,5-trifluorophenyl)butanoyl]-3-[(2-methylpropan-2-yl)oxymethyl]piperazin-2-one;(2R,3R)-2,3-dihydroxybutanedioic acid (Fig. 1) [8–10]. It is marketed as VALERA generic name coined by Alkem Laboratories, for the treatment of type-II diabetes mellitus as a monotherapy or in combination with metformin HCl. The usual adult dosage is 5 mg of evogliptin tartrate administered orally once daily. It is available as white amorphous powder, practically soluble in water, methanol and exhibits high oral bioavailability owing to extensive hepatic first-pass effect, and CYP3A4 plays the major role in hydroxylation of evogliptin to M7 and M8, and UGT2B7 plays the major role in glucuronidation of M7 to M16 [11].

Molecular structure of evogliptin tartrate
According to the literature reviews, few analytical methods [12,13,14,15,16] such as UV [17, 18] and HPLC have been reported for the estimation of evogliptin tartrate in bulk and pharmaceutical dosage form but no one has used quality by design approach. The major objective of AQbD has been identified failure modes and established robust method with operable design space throughout the product life cycle management [19, 20]. The QbD method was based on the understanding and implementation of guideline ICH Q8 pharmaceutical development, ICH Q9 quality risk management and ICH Q10 pharmaceutical quality system [21,22,23].
The current work intends to develop and optimize the RP-HPLC method by employing analytical QbD principles for the estimation of evogliptin tartrate pharmaceutical dosage form. Implementation of QbD principle for analytical method development has been practised quite popular for attaining the high robustness and enhance method performance. The QbD method was based on the understanding and implementation of guideline ICH Q8 pharmaceutical development, ICH Q9 quality risk management and ICH Q10 pharmaceutical quality system.
Methods
Chemicals
The evogliptin tartrate sample was provided by Alkem Laboratories Limited, Sikkim, India. The commercially available tablet formulation of Valera (evogliptin tartrate 5 mg) was used for assay. HPLC grade methanol and water were purchased from Merck, Mumbai, India, for the study.
Instrumentation and chromatographic conditions
A Shimadzu high-performance liquid chromatography system fitted with quaternary solvent manager, a solvent degasser, sample manager, PDA detector controlled by LC solution software, cooling autosampler and column oven enabling temperature control of the analytical column was used for the method development. All precision weighing was performed on Shimadzu analytical balance. Chromatographic separation was performed on a reverse phase water C18 column (150 mm × 4.6 mm) having 5.0-μm particle size. Isocratic elution was used with methanol:phosphate buffer (pH 4.5) in the ratio 60:40% v/v as a mobile phase. The flow rate kept at 1 ml/min, and elutes were supervised using PDA detector at 267 nm. Design Expert 13.0 trial version was used for designing experiments.
Preparation of standard stock solution
Stock solution of evogliptin tartrate (1000 μg/ml) was prepared by dissolving accurate quantity of 25 mg of evogliptin tartrate in 25 ml of methanol. Using this stock solution, various working solutions (10–60 μg/ml) were prepared by the serial dilution with methanol. The prepared all solution were filtered through a nylon filter (0.45 μ) and subjected to chromatographic analysis for preparing the calibration plot.
Preparation of sample solution
Accurately weighed equivalent quantity to 25 mg of evogliptin tartrate in a 25 ml of volumetric flask. Transfer 15 ml of methanol into flask and shake well properly then sonicate for 15 min. The flask was shaken and made up to the mark with diluent. The above solution was filtered through Whatman filter paper (0.45 μ). The solution was made up to the mark with diluent to give a solution containing 20 μg/ml of EVO and analysed it by proposed method.
Selection of detection wavelength
EVO standard solution was scanned in the range of 200–400 nm, and wavelength maxima 267 nm were selected as a detection wavelength.
Force degradation study
Forced degradation is a procedure that includes the degradation of drug molecule at different stress conditions that can be used to find out the stability of the drug substance.
Acidic and alkali degradation
For acidic and alkali degradation, stock solution of EVO was taken into separate flasks and added 0.1 N HCl and 0.1 N NaOH, respectively; then, the volumetric flasks were kept at 60 °C in reflux condition 1 h. For oxidation degradation study, transferred the stock solution of EVO into separate flask and added 30% v/v of hydrogen peroxide into it then volumetric flask was kept in dark place for 3 h. For thermal study, standard EVO powder was placed in an oven at 100 °C for 24 h. For photostability degradation, standard EVO powder was placed in UV chamber, and the drug was exposed to UV rays for 72 h. All samples were diluted to obtain a final concentration of 20 μg/ml of EVO.
Method development through QbD approach
Defining the quality target profile
For analytical method, development APT (analytical target profile) plays an important role for identifying the variables that are needed to ensure the quality characteristics and purpose of the analytical method. Analyst, sample and various analytical techniques are the part of target profile for efficient method development.
Determine critical quality attributes (CQA)
In AQbD analysis, CQAs are selected on the basis of which techniques used for method development (RP-HPLC) and method intent (e.g. assay and drug release determination). Tailing factor, theoretical plate count, resolution, retention time and % recovery are the CQAs for the assay determination method.
Identify critical method parameters (CMP) and risk assessment of method
CMPs are the sensitivities associated with the analytical method. CMPs have a direct relation with CQAs. CMPs can be classified on the basis of different analytical techniques such as GC, HPLC and HPTLC. For RP-HPLC method, % organic modifier in mobile phase, pH of mobile phase, column temperature, flow rate and injection volume are the various CMPs for the method development.
Risk assessment provides probable interactions with CMPs and assessing the probabilities of successive failure. An Ishikawa fishbone diagram was sketched to determine the potential risks, and root causes that effect on the performance of RP-HPLC.
Design of experiment (DOE)
For DOE selection, response surface design was selected with compared to factorial design. Box–Behnken was chosen for further study because this study gave good interaction with three-factor and three-level design with minimum runs as compared to central composite design. On the basis of factor screening studies, the method performance was optimized using Box–Behnken response surface design at three levels, i.e. low (− 1), intermediate (0) and high (+ 1). Selected CMPs factors (% organic modifier, flow rate and pH of buffer) are selected as an input variable while CQAs (retention time and tailing factor) are selected as a response variable in DOE (Table 1).
A design matrix listing 15 experimental runs was generated using Design Expert tools. The various interaction effect and quadratic effects of the % organic modifier, pH and flow rate on the retention time and tailing factor were studied using Box–Behnken screening design (Table 2).
Analytical method development
The developed method was validated as per ICH guidelines for linearity, accuracy, precision, specificity and robustness, limit of detection and limit of quantification [23].
Linearity
Linearity of the developed method was determined by analysing the serial dilution of stock solution of EVO between 10 and 60 μg/ml and plotting the calibration curve of peak area against concentration of EVO. Linearity of the method was confirmed by the regression statistics analysis on the obtained data using MS Excel spreadsheet.
Precision
Precision of the developed method was assessed by the three different concentrations of EVO (20 μg/ml, 30 μg/ml and 40 μg/ml) at different time intervals on the same day (intraday precision) and by consequent days (\iInterday precision). Calculate the % RSD of the all data.
Accuracy
Accuracy was determined by addition method. Sample solution of EVO (20 μg/ml) was spiked with 80% (16 μg/ml), 100% (20 μg/ml) and 120% (24 μg/ml) of the std. EVO. Further, recovery and % RSD were calculated to check the accuracy of data.
Limit of detection (LOD) and limit of quantification (LOQ)
LOD and LOQ were determined from the slope of calibration plot and standard deviation of the response to the blank sample.
Formula of LOD and LOQ is,
where σ = standard deviation ands = slope of calibration curve.
Robustness
Robustness of the method was determined by deliberate change in the system suitability parameters, i.e. alteration volume of mobile phase, flow rate and injection volume. A working solution 20 μg/ml was used for all experimental run of robustness parameter.
System suitability
The system suitability was determined by six replicate analyses of std. EVO (20 μg/ml) followed by estimation of SD and % RSD of peak area, tailing factor, retention time and theoretical plate.
Results
Method development studies
As per the literature reports, the developing LC method describes the use of complex mobile phase with different compositions (containing solvents such as methanol, water, acetonitrile and phosphate buffer at a particular pH), variable flow rate (isocratic/gradient), column (C8 and C18), temperature setting, etc., for chromatographic separation.
The preliminary studies suggested the selection of methanol and phosphate buffer (pH 4.5) as suitable mixtures for the mobile phase along with adequate peak symmetry and lower peak tailing.
Optimization of HPLC method by QbD approach
The analytical target profile selected was retention time, tailing factor and theoretical plates for optimization of HPLC conditions. The Box–Behnken design was used for further the optimization of various parameters within the design space. The data analysis was carried out by selecting the quadratic model for main and interaction effects. The optimized model parameters are shown in Table 3. The model generates quadratic equation for each CAA parameter. Equation for retention time (for actual value) = 148.65 − 2.30 A − 26.81 B − 24.83 C + 0.069 A * B + 0.16 A * C + 0.31 B * C + 0.15 A2 + 2.45 B2 + 5.36 C2. It was concluded that as β1 negative coefficient (− 2.30) suggests that the amount of mobile phase (A) decreases and β2 negative coefficient (− 26.81) suggests that as pH of buffer (B) decreases, and β3 negative coefficient (− 24.83) suggests that the flow rate (C) decreases, the value of retention time was increased (Fig. 2).

Three-dimensional surface plot for the effect of flow rate and pH on retention time
Equation for tailing factor (for actual value) = 4.50 − 0.108 A + 0.019 B + 0.25 C − 0.003 A * B − 0.007 A * C − 0.15 B * C + 0.001 A2 + 0.035 B2 + 0.39 C2. It was concluded that as β1 negative coefficient (− 0.108) suggests that the amount of mobile phase (A) decreases and β2 positive coefficient (+ 0.019) suggests that as pH of buffer (B) increases, and β3 negative coefficient (+ 0.25) suggests that the flow rate (C) increases, the value of tailing factor was increased (Fig. 3).

Three-dimensional surface plot for the effect of flow rate and pH on tailing factor
Equation for theoretical plate (for actual value) = − 15,518.37 + 1201.6 A − 2571.7 B − 29,593.7 C − 95.1 A * B + 165 A * C + 840 B * C − 7.69 A2 + 838.5 B2 + 10,596.8 C2. It was concluded that as β1 positive coefficient (+ 1201.6) suggests that the amount of mobile phase (A) increases and β2 negative coefficient (− 2571.7) suggests that as pH of buffer (B) decreases, and β3 negative coefficient (− 29,593.7) suggests that the flow rate (C) decreases, the value of theoretical plate was increased (Fig. 4).

Three-dimensional surface plot for the effect of flow rate and pH on theoretical plate
The optimized solution showed the mobile phase composition containing 60:40% v/v mixture of methanol:phosphate buffer pH (4.5) and flow rate 1 ml/min yielded desirability close to 1.0 along with all the CAAs in the desired range (Table 4).
Method validation studies
Linearity
The linear calibration plot for EVO in concentration range between 10 to 60 μg/ml showed good linearity with higher value of correlation coefficient of 0.998 (Fig. 5, Table 5).

Calibration curve of evogliptin tartrate
Accuracy
Accuracy data showed good percentage recovery between 98.68 to 101.06% at all three levels and % RSD value not more than 2% which indicates high degree of accuracy of developed method. Result and statistical data of accuracy are given in Table 6.
Precision
The % RSD of intraday and interday precision for EVO on three times measurements was found to be less than 2%. These results confirmed high degree of precision of the developed method. Table 7 illustrates the precision data of method.
Limit of detection and limit of quantification
The limit of detection (LOD) and limit of quantification (LOQ) data for EVO as 0.691 μg/ml and 2.095 μg/ml thus indicate very high sensitivity of the developed method for quantification of EVO.
System suitability
System suitability data show the lack of significant difference in the peak area, retention time, theoretical plate and tailing factor of EVO with six replicates. The % RSD of all system suitability parameters were found to be less than 1% (Fig. 6a and b).

a Chromatogram of standard evogliptin tartrate (20 μg/ml) and b chromatogram of sample evogliptin tartrate (20 μg/ml)
Robustness
The robustness was studied by the slight but deliberate change in parameters such as pH of mobile phase, flow rate and injection volume. The % RSD of all parameters were found to be within limit, i.e. less than 2%.
Applicability of developed method
The % assay of drug content was found to be 99.81% (n = 6) for label claim of EVO. The assay result indicates selectivity of the developed RP-HPLC method for the estimation of EVO in pharmaceutical dosage form (Table 8).
Force degradation study
The standard solutions were applied different stress conditions mentioned in the procedure. The drug showed 12.89% degradation with 1 degraded peak in acidic conditions (Fig. 7). Under alkali condition, the drug showed 17.36% degradation with 2 degraded peak (Fig. 8). Under oxidation condition, the drug showed 12.64% degradation, and 1 degraded peak was observed (Fig. 9). In thermal and photolytic condition, the drug showed 11.15% and 10.45% degradation, respectively, and no degraded peak was observed (Figs. 10 and 11).

Chromatogram of EVO (20 µg/ml) under acidic degradation condition

Chromatogram of EVO (20 µg/ml) under alkaline degradation condition

Chromatogram of EVO (20 µg/ml) under oxidative degradation condition

Chromatogram of EVO (20 µg/ml) under thermal degradation condition

Chromatogram of EVO (20 µg/ml) under photolytic degradation condition
Discussion
The present study was to create a simple, accurate, reliable and successful application of QbD principles for the development of RP-HPLC method for the estimation of evogliptin tartrate with improved robustness and performance. The Box–Behnken design was applied for three factors at three levels with use of the Design Expert software trial version 13.0. Design of experimental results, including ANOVA, diagnostic graphs and model graphs, were examined for each factor. The effect of each factor on the response result was investigated in this results part. The developed method was applied for degradation study and adequately separated the drug with its degradation product. EVO was significantly degraded in acidic, alkali and oxidative conditions. The established RP-HPLC method was validated in terms of linearity, precision, accuracy, robustness, LOD and LOQ as per ICH Q2 R1 guidelines. Applicability of developed method for quantification of EVO in pharmaceutical formulations shown the absence of any wanted peak and no change in retention time of the drug, thus high specification and selectivity of the method is fulfil.
Conclusion
A simple, rapid, sensitive and economical analytical method has been successfully developed employing the QbD-based approach for the estimation of EVO in bulk and pharmaceutical dosage form. A detailed statistical analysis and design spaces are established through DOE for the developed method, and method is proved to be robust for wider variation in conditions. It can be effectively applied for routine analysis in research institutions, quality control department in industries and approved testing laboratories.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Abbreviations
- EVO:
-
Evogliptin tartrate
- QbD:
-
Quality by design
- ICH:
-
International Conference on Harmonization
- RP-HPLC:
-
Reverse-phase high-performance liquid chromatography
- ANOVA:
-
Analysis of variance
- DOE:
-
Design of experiments
- µg/ml:
-
Microgram per milliliter
- PDA:
-
Photodiode array
References
Tan X, Hu J (2016) Evogliptin: a new dipeptidyl peptidase inhibitor for the treatment of type-2 diabetes. Expert Opin Pharmacother 17(9):1285–1293
Thakkar K, Patel K et al (2021) Evogliptin a new drug of DPP-4 inhibitor: an overview. World J Pharm Res 5(10):1921–1929
Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M (2015) Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 38(1):140–149
Kim M-J, Kim N-Y et al (2018) Evogliptin, a dipeptidyl peptidase-4 inhibitor, attenuates renal fibrosis caused by unilateral ureteral obstruction in mice. Diabetes Meta J 43(1):271–278
Rodbard HW, Jellinger PS, Davidson JA (2009) Statement by an American Association of Clinical Endocrinologists American college of Endocrinology consensus penal on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract 15(6):540–559
Seo S, Kim MK, Kim RI, Yeo Y, Kim KL, Suh W (2021) Evogliptin, a dipeptidyl peptidase-4 inhibitor, attenuates pathological retinal angiogenesis by suppressing vascular endothelial growth factor-induced Arf6 activation. Exp Mol Med 52(10):1744–1753
Chae YN, Kim TH, Kim MK, Shin CY, Jung IH, Sohn YS (2015) Beneficial effects of Evogliptin, a novel dipeptidyl peptidase 4 inhibitor, on adiposity with increased Ppargc1a in white adipose tissue in obese mice. PLoS ONE 10(12):e0144064
Drug bank (DB12625) (2021) Evogliptin. https://go.drugbank.com/drugs/DB12625. Accessed 15 Mar 2021
PubChem (135395528) (2021) Evogliptin tartrate. https://pubchem.ncbi.nlm.nih.gov. Accessed 15 Mar 2021
Alkemlabs (2021) Evogliptin. https://www.alkemlabs.com/pdf/adverse/Evogliptin.pdf. Accessed 15 Mar 2021
Lee DY, Kim JH, Shim HJ, Jeong HU, Lee HS (2018) Absorption, metabolism, and excretion of [14C] evogliptin tartrate in male rats and dogs. J Toxicol Environ Health 81(11):453–464
Shah UA (2021) A stability indicating RP-HPLC method validation for simultaneous estimation of metformin HCl and canagliflozin in pharmaceutical dosage form. J Pharm Res 33(56A):180–192
Shah UA (2021) A stability indicating RP-HPLC method validation for simultaneous estimation of metformin HCl, dapagliflozin and saxagliptin in pharmaceutical dosage form. J Pharm Res 33(59A):754–767
Patel K, Dr.Ujashkumar A. Shah, (2022) Qbd stressed development and validation of stability-indicating RP- HPLC method for the simultaneous estimation of linagliptin and metformin HCl in pharmaceutical dosage form. Res J Pharm Technol 15(5):1917–1923
Patel S, Jagtap K, Shah U, Patel D (2022) Development of validated stability-indicating chromatographic method for the determination of metformin and teneligliptin and its related impurities in pharmaceutical tablets. Int J Pharm Qual Assur 13(2):128–136
Sanyal AP, Patel K (2021) Analytical method development for estimation of saroglitazar magnesium in pharmaceutical dosage form by quality by design approach. World J Pharm Pharm Sci 9(4):1267–1281
Shah PB, Patel K (2021) Qbd—a novel setup for the analytical method development. World J Pharm Pharm Sci 10(5):1645–1662
Patel K, Shah UA, Joshi H, Patel CN (2023) Derivative spectrophotometric method development and validation for the estimation of evogliptin tartrate in pharmaceutical dosage form. Indian J Pharm Educ Res 57(1):228–233
Sharma D, Patel K, Patel CN (2021) QbD-a new era of pharmaceutical drug development. Eur J Pharm Med Res 8(5):688–694
ICH harmonized tripartite guidelines, pharmaceutical development: text and methodology, Q8, Geneva, Aug 2009
ICH harmonized tripartite guidelines, quality risk management: text and methodology, Q9, Geneva, Nov 2005
ICH harmonized tripartite guidelines, pharmaceutical quality systems: text and methodology, Q10, Geneva, June 2008
ICH harmonized tripartite guidelines, validation of analytical procedures: text and methodology, Q2 (R1), Geneva, Nov 2005
Acknowledgements
The authors are highly thankful to Dr. Ujash shah for your guidance and motivation support. The authors are thankful to Dr. C. N. Patel, Principal of Shri Sarajanik Pharmacy College, Gujarat, India, for the providing all the facilities to carry out the research work.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
KP performed analysis and validation parameters and prepared the manuscript. UAS interpreted analytical data and helped in design expert software handling. Dr. CNP provided facilities and helped in the project performance. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The authors declare no conflict of interest.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Patel, K., Shah, U.A. & Patel, C.N. Box–Behnken design-assisted optimization of RP-HPLC method for the estimation of evogliptin tartrate by analytical quality by design. Futur J Pharm Sci 9, 57 (2023). https://doi.org/10.1186/s43094-023-00509-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-023-00509-w