
Key-points
Bone turnover makers (P1nP, B-ALP, CTX), bone modulators (Dkk1, sclerostin) and BMD were measured prospectively in rheumatoid arthritis patients treated with tofacitinib.
Sclerostin increased significantly after treatment with tofacitinib, P1nP and B-ALP (markers of bone formation) decreased significantly.
Abstract
Background
Rheumatoid arthritis (RA) is characterized by bone loss. It is unclear whether JAK inhibitors can attenuate bone loss in RA by modulating bone metabolism. The main objective of our study is to investigate the effects of tofacitinib on serum levels of bone turnover markers and modulators. Secondary objectives were to assess changes in bone mineral density (BMD), metacarpal index, bone erosions.
Methods
We conducted a prospective observational study on patients with active RA failure to bDMARDs or tsDMARDs initiating treatment with tofacitinib. We measured at baseline and after 1, 2, 3, 6, 9 and 12 months: serum bone turnover markers (CTX, P1nP, B-ALP), bone modulators (Dkk-1, sclerostin, vitamin D, PTH, OPG and RANKL), BMD and radiographic parameters (Sharp van der Heijde score [SvdH], bone health index [BHI] and metacarpal index [MCI]).
Results
30 patients were enrolled in the study of whom 21 completed the study through month 12. Tofacitinib was clinically effective by suppressing DAS28-CRP. Glucocorticoids daily dose significantly decreased from baseline. We found a negative correlation between pre-study cumulative and daily dose of glucocorticoids and baseline B-ALP serum levels (r -0.592, p 0.012). Sclerostin serum levels increased significantly during the study period, while P1nP and B-ALP (markers of bone formation) decreased significantly. BMD levels, BHI, MCI and SvdH score did not change.
Conclusion
Treatment with tofacitinib was associated with a significant increase in sclerostin serum levels and a parallel decrease in markers of bone formation. However, no significant bone loss was observed.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by inflammation, leading to joint destruction and functional impairment [1]. In addition to the articular manifestations, RA is associated with several systemic complications, including osteoporosis and an increased risk of fractures [2]. While glucocorticoid intake and comorbidities partially contribute to this heightened risk of bone loss [3], there are other metabolic features of RA that are thought to play a significant role [4, 5]. Bone remodeling, a complex process involving bone formation and resorption, is regulated by various signaling pathways. The Wnt signaling pathway has emerged as a key regulator of joint and bone remodeling, and its dysregulation has been implicated in the pathogenesis of bone loss in RA [6, 7]. In particular, inhibitors of the Wnt pathway, such as sclerostin and Dickkopf-1 (Dkk1), have been found to be dysregulated in RA patients compared to controls and other inflammatory rheumatic musculoskeletal diseases (iRMDs) [8,9,10]. However, it remains unclear whether disease-modifying antirheumatic drugs (DMARDs) can directly modulate the metabolic profile of RA, specifically targeting bone cells and bone metabolism [11].
Tofacitinib, a Janus kinase (JAK) inhibitor, has demonstrated efficacy in the management of RA by effectively controlling inflammation [12]. While previous studies have investigated the effects of targeted therapies, particularly tumor necrosis factor (TNF) inhibitors, on bone remodeling in RA, limited research has explored the impact of JAK inhibition on bone remodeling or fracture risk [11]. A notable exception comes from a pooled data analysis of randomized clinical trials investigating the effects of tofacitinib, which revealed a significant higher incidence of fractures when compared to TNF inhibitors users [13].
The primary objective of our study was to investigate the impact of tofacitinib on bone turnover markers in RA patients. Understanding the effects of tofacitinib on bone remodeling in RA patients is crucial for optimizing treatment strategies and mitigating the risk of osteoporosis and fractures. By elucidating the direct effects of tofacitinib on bone turnover markers, we aim to contribute to a comprehensive understanding of the metabolic alterations associated with RA and its therapeutic interventions.
Materials and methods
We did a prospective observational analysis of patients suffering from active RA, who had not responded to targeted synthetic or biological Disease Modifying Anti-Rheumatic Drugs (tsDMARDs or bDMARDs), and who had been on a stable dose of glucocorticoids of ≤ 5 mg/day of prednisone equivalent for at least three months, beginning treatment with tofacitinib 5 mg twice a day. The observation period lasted 12 months, during which patients underwent scheduled control examinations and study-related assessments at the outset (Baseline, B), and then at month 1 (M1), month 2 (M2), month 3 (M3), month 6 (M6), month 9 (M9), and month 12 (M12). The study document outlines all procedural details. Adjustments to the dose of conventional DMARDs (csDMARDs) and a short course of glucocorticoids (exceeding 5 mg/day of prednisone equivalent for no more than 15 days) were allowed, though any changes were meticulously documented. In alignment with the study guidelines, while efforts were made to maintain consistency in the administration of DMARDs and glucocorticoids, adjustments to ensure the best possible clinical care for patients were permitted, including the reduction of glucocorticoid dosage. Clinical trial number: not applicable.
Inclusion and exclusion criteria
Eligibility criteria included: being 18 years or older, providing signed informed consent, having a RA diagnosis as per the ACR/EULAR 2010 criteria, disease manifestation within the past three years, necessity for tofacitinib treatment owing to ineffectiveness or intolerance to tsDMARDs or bDMARDs, maintenance of a stable glucocorticoid dose of ≤ 5 mg/day of prednisone equivalent for a minimum of three months before joining the study, and a sufficient clearance period from previous bDMARDs or tsDMARDs, defined as at least three half-lives of the biologic agent or a minimum of three months. Non-eligibility criteria comprised: having a rheumatic diagnosis other than RA, suffering from bone conditions aside from osteoporosis (for example, Paget’s disease of bone), advanced liver or kidney disease (eGRF < 30 ml/min or Child-Pugh grade B or C), untreated endocrine disorders, contraindications to tofacitinib use as indicated on its label, undergoing treatments with bisphosphonates (with a grace period up to 12 months for oral bisphosphonates and up to 24 months for zoledronic acid), strontium ranelate, teriparatide, selective estrogen receptor modulators (SERM), or denosumab, receiving intra-articular injections in the MCPs or MTPs within three months prior to the study, or being pregnant or breastfeeding.
Sample size
This was an exploratory biomarker development study, the sample size of 30 patients was based on clinical judgement and practical considerations and not on formal statistical reasoning.
Study procedures
Before joining the study, participants underwent a screening process to ensure they met the necessary inclusion and exclusion criteria and then attended their initial visit. During each visit (initial, M1, M2, M3, M6, M9, and M12), we conducted clinical evaluations, reviewed the safety profile, assessed ongoing treatments for RA (including glucocorticoids and csDMARDs doses), and evaluated any other medications being taken. Additionally, we calculated the DAS28-CRP score.
Bone Mineral Density (BMD) was assessed at the femoral neck and lumbar spine (L1-L4) areas using dual-energy X-ray absorptiometry (DEXA) (utilizing the QDR Hologic Delphi system) during the initial visit, M6, and M12. The coefficient of variation was set at 1% for the vertebral site and 1.2% for the femoral neck.
Standard X-rays of the hands and feet were taken at the beginning and end of the study (M12). The modified Sharp/van der Heijde score (mSvdH score) was determined by two separate experienced evaluators. Hand X-ray images, provided in DICOM format, were examined using the Bonexpert for Adults software by Visiana Version 2.3.0.4, which produced the Bone Health Index (BHI) and the Metacarpal Index (MCI), both indicators of cortical bone thickness derived from the dimensions of the three central metacarpal bones. We calculated the average BHI and MCI values for both the right and left hands.
Blood samples were collected during fasting in the morning at the initial visit, M1, M2, M3, M6, and M12, then aliquoted and preserved at − 80 °C for future analysis. We previously described the analysis of biomarkers in a previous study [14]. In synthesis we tested: CTX, P1nP, B-ALP, Sclerostin, Dkk-1, PTH, vitamin D, RANKL and OPG.
Statistical analysis
Comparisons between groups were conducted using the t-test and the Mann–Whitney U test for data that followed a normal distribution and data that did not, respectively. The chi-square (χ2) test was utilized for analyzing categorical data. To examine the relationship between continuous variables, we employed Pearson correlation coefficients. The False Discovery Rate (FDR) method, specifically the two-stage step-up procedure by Benjamini, Krieger, and Yekutieli (with an FDR Q value of 5%), was applied to address the issue of multiple comparisons.
For analyzing changes in Bone Mineral Density (BMD), serum markers, and clinical measures over time, we used a mixed-effects model for repeated measures (MMRM). In instances of missing data, traditional repeated measures ANOVA is not applicable. Hence, we adopted the use of GraphPad Prism version 9.5.1 to fit a mixed model that applies a compound symmetry covariance matrix, utilizing the Restricted Maximum Likelihood (REML) method. Notably, this method yields identical p-values and multiple comparison tests as repeated measures ANOVA when there are no missing values. For multiple comparisons adjustments, we applied Tukey’s procedure to correct p-values. The differences in SvdH scores, BHI, and MCI for paired groups were assessed using the Wilcoxon Signed-Ranks Test. To adjust for the potential influence of disease activity (both at baseline and follow-ups) we conducted an ANCOVA (GLM) with markers that were found to significantly change as dependent variable and including DAS28 at baseline and deltaDAS28 at M12 as covariates.
All differences were considered significant when p value was inferior to 0.05. All statistical analyses were performed using SPSS Version 26 (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism version 9.5.1 (GraphPad Software, San Diego, CA, USA). This study was approved by the University of Verona ethic committee (prot. WI232128).
Results
Patients characteristics
Thirty patients (86.7% women) that satisfied inclusion criteria were consecutively enrolled in the study. Mean age was 56.1 years (SD 10.4), mean disease duration was 1.5 years (SD 0.6), 13 patients (43.3%) had seropositive RA. Twenty-four patients (80.0%) were concomitantly treated with csDMARD (21 with methotrexate, 2 with leflunomide and 1 with sulfasalazine). None of the patient ever received an anti-osteoporosis medication other than vitamin D supplements. Patients baseline characteristics are given in Table 1. One patient left the study at M6 (adverse events to tofacitinib) and other 6 patients left the study at M9 (1 for inefficacy and 5 lost in follow-up). Complete data analysis was available for 23 patients in whom the baseline characteristics did not differ significantly from the overall population (data not shown). Disease activity significantly decreased from baseline (mean DAS28-CRP 4.09, SD 1.59) to M12 (mean DAS28-CRP 2.18, SD 1.04), p for linear trend < 0.001. Similarly, GC daily dose significantly decreased from baseline (mean dose 3.3 mg, SD 4.2) to M12 (mean dose 1.4 mg, SD 2.8) p for linear trend 0.012.
mSvdH score, BHI and MCI
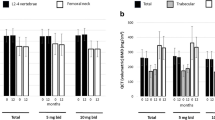
mSvdH score did not change form baseline to M12 (median score 23.0, IQR 12.0–60.0 and 21.0, IQR 13.0–54.0 at baseline and M12 respectively, p 0.355). BHI and MCI did not change from baseline to M12 as well (mean BHI 5.4, SD 0.7 and 5.2, SD 0.7 at baseline and M12 respectively, p 0.193 and mean MCI − 0.36, SD 1.00 and − 0.35, SD 1.18 at baseline and M12 respectively, p 0.431). SvdH score, BHI and MCI changes are depicted in Fig. 1 (upper panel).

Changes in Sharp van der Heijde (SvdH) score, metacarpal index (MCI) and bone health index (BHI) in the upper panel. Changes in bone mineral density (BMD) at lumbar spine, total hip and femoral neck in the lower panel. BMD changes were tested with mixed-effect model analysis for repeated measures (MMRM), p-values are adjusted for multiple comparisons using the Tukey’s procedure. SvdH score, BHI and MCI changes were tested with Wilcoxon signed-ranks test. Error bars represent 95% CI
Bone mineral density
Average BMD levels at baseline were: 0.922 (SD 0.108), 0.766 (SD 0.120) and 0.911 (SD 0.151) at lumbar spine, femoral neck and total hip, respectively. At baseline 2 (6%) patients classified as osteoporotic (T-score ≤-2.5) and 16 (53%) classified as osteopenic (T-score ≤-1.0 and >-2.5) at any site. BMD (both at lumbar spine, femoral neck and total hip) at baseline did not correlate with any of the patients’ characteristics or baseline markers’ serum levels (p adjusted for multiplicity NS). BMD did not change significantly throughout the study period. BMD changes are shown in Fig. 1 (lower panel). At month 12 BMD, T-score and Z-score levels were as follows: lumbar spine BMD 0.927 (SD 0.132), T-score − 1.15 (SD 1.13), Z-score − 0.06 (SD 1.14); femoral neck BMD 0.739 (SD 0.128), T-score − 0.81 (SD 0.76), Z-score 0.20 (SD 0.13); total hip BMD 0.954 (SD 0.134), T-score 0.02 (SD 1.04), Z-score 0.66 (SD 1.00). At month 12, 2 (9% of 23) patients classified as osteoporotic (T-score ≤-2.5) and 10 (43% of 23) classified as osteopenic (T-score ≤-1.0 and >-2.5) at any site.
Bone turnover markers
At baseline we found a negative correlation between cumulative and daily dose of glucocorticoids and B-ALP (r -0.592, p 0.012 corrected for multiplicity).
In Fig. 2 are shown the changes of bone turnover markers in the study period. Serum CTX remained stable over the time. B-ALP decreased significantly at M6 while P1nP serum levels decreased significantly at M6 and M12, respectively. In the ANCOVA corrected for disease activity modifications we did not find any interaction (p value 0.345 for time*deltaDAS28 interaction) and the pairwise comparisons were not modified substantially.

Changes in bone turnover markers (BTMs) in the study period. Changes were tested with mixed-effect model analysis for repeated measures (MMRM), p-values are adjusted for multiple comparisons using the Tukey’s procedure. Error bars represent 95% CI
Bone metabolism modulators
In Fig. 3 are shown the changes in Dkk1, sclerostin, RANKL, OPG, vitamin D and PTH serum levels in the study period. Among bone metabolism modulators sclerostin increased significantly during the study period (p trend 0.042). The increase was consistently found between baseline and M1 (p 0.044), M2 (p 0.020), M3 (0.005), M6 (p 0.003) and M12 (p0.029); p values adjusted for multiple comparisons. Results did not change when we controlled for time varying glucocorticoid use, time varying DAS28, age, sex and seropositivity. In detail, in the ANCOVA corrected for time varying disease activity we did not find any interaction (p value 0.345 for time*deltaDAS28 interaction) and the pairwise comparisons were not modified substantially. All other bone modulators did not change. When accounting for multiplicity, we did not find any other significant correlation between baseline levels of bone turnover markers or bone metabolism modulators and clinical/densitometric/radiographic parameters. Similarly, we did not find any significant correlation between changes in such parameters. Figure 4 shows the correlation matrix between baseline characteristics and bone parameters in the study population, p-values are adjusted for multiplicity using the two-stage step-up procedure by Benjamini, Krieger, and Yekutieli (with an FDR Q value of 5%).

Changes in serum bone modulators in the study period. Changes were tested with mixed-effect model analysis for repeated measures (MMRM), p-values are adjusted for multiple comparisons using the Tukey’s procedure. Error bars represent 95% CI

Correlation matrix of baseline characteristics and bone parameters of the study population. Asterisks shows p-value < 0.05 adjusted for multiplicity using the two-stage step-up procedure by Benjamini, Krieger, and Yekutieli (with an FDR Q value of 5%)
Discussion
We conducted an observational longitudinal study on RA patients receiving tofacitinib. In aggregate we found that tofacitinib caused a significant increase in sclerostin, a Wnt inhibitor, and a small, but significant, decrease in makers of bone formation (B-ALP and P1nP).
RA has been largely associated with osteoporosis and fracture risk, which seems to be only partially related to glucocorticoid intake and associated comorbidities [2, 15]. Indeed, such higher risk of systemic bone loss is also explained by several metabolic features of RA [4]. For example, Dkk1, which is widely considered as the master regulator of joint and bone remodeling, has been consistently found elevated in RA as compared to controls and other iRMDs. However, it is unclear whether DMARDs can actually modify the metabolic profile of RA acting directly on bone cells and bone metabolism. Indeed, albeit many studies have been conducted exploring the effects of targeted therapies in RA, the role of such medications on biomarkers of bone remodeling is still unclear [11]. Furthermore, many of these studies have been conducted on TNF or IL6 inhibitor users, limiting our knowledge on other mechanisms of action. For example, the effect of JAK inhibition on bone turnover markers has been explored only in one study by Hamar and colleagues [16]. The authors found that tofacitinib treatment (pooling 5 mg bis in die and 10 mg bis in die) triggered a significant decrease in CTX serum levels while increasing serum osteocalcin. This result is in direct opposition to our results on bone turnover markers. Indeed, we found a significant decrease in bone anabolic markers contrasting the results by Hamar et al.
While our study and the study conducted by Hamar and colleagues share similarities in design, it is important to highlight several significant differences between them. First, approximately 50% of the patients enrolled in Hamar’s study were treatment-naïve, whereas we specifically included patients who had failed to respond to at least one bDMARD or tsDMARD. While enrolling treatment-naïve patients may be advantageous from a metabolic perspective, as it minimizes potential disturbances in bone metabolism, it may limit the generalizability of the results since tsDMARDs are often prescribed as second-line treatments. Second, in our study, we allowed for glucocorticoid tapering while keeping the dose as stable as possible due to obvious ethical considerations. In contrast, the subjects in Hamar’s study did not receive glucocorticoids during the study period or in the three months leading up to the study, despite experiencing high disease activity at baseline. This difference in glucocorticoid use may have significantly influenced the markers of bone remodeling, but it also implies a greater generalizability of our findings. Notably, we observed a negative correlation between levels of B-ALP, a marker of bone formation, and cumulative glucocorticoid dose, further highlighting the prominent impact of steroids on bone markers. Third, Hamar and colleagues reported a significant increase in 25OHvitD levels despite claiming a stable dose of vitamin D supplements during their study, with approximately half of the cohort taking vitamin D supplements. Specifically, they observed an average increase from 16 ng/mL to over 24 ng/mL in patients treated with tofacitinib 5 mg twice daily (BID). In contrast, we did not observe any appreciable changes in serum 25OHvitD levels, and all but one patient consistently took vitamin D supplements throughout the duration of the study. Fluctuations in vitamin D levels have been shown to greatly impact bone turnover markers as well as disease activity and inflammation in RA, as supported by previous research [17,18,19]. These discrepancies in vitamin D levels may have contributed to the divergent results observed by Hamar et al. Lastly, Hamar and colleagues utilized osteocalcin as a marker of bone formation, whereas we employed P1nP, which is widely recognized as a preferable marker for assessing bone neo-apposition [20]. The choice of bone formation marker can influence the interpretation of results, and P1nP is considered a more reliable indicator of bone formation activity.
By acknowledging these important differences between our study and that of Hamar et al., we aim to provide a comprehensive understanding of the complexities involved in evaluating the effects of tofacitinib on bone remodeling in patients with RA. Each study design and approach bring unique strengths and limitations, which should be carefully considered when interpreting and generalizing the results.
The JAK-STAT pathway is one of the numerous cytokines signaling pathways found in bone cells [21]. To our knowledge, our is the first study investigating the effects of a JAK inhibitor on the Wnt system in RA patients. In our study tofacitinib treatment produced a significant increase in sclerostin serum levels while Dkk1, another Wnt system inhibitor, as well as PTH, RANKL and OPG remained unchanged. Our finding was consistent even controlling for disease activity and other covariates known for inducing metabolic disturbances (glucocorticoids, seropositivity etc.). Sclerostin increase might well explain the drop in bone formation markers we noticed. Sclerostin is indeed a potent inhibitor of the Wnt system which is the main stimulator of osteoblast activity; therefore, the P1nP and B-ALP decrease was somehow expected. This result might appear controversial since it has been shown that tofacitinib can significantly increase osteoblast function in animal models, resulting in prominent bone anabolism and repair of bone erosions [22]. Nonetheless, our findings are in line with a recent observation from a sub analysis of the ORAL-surveillance trial [13]. In this large randomized controlled trial, the risk of osteoporotic fracture was significantly greater in tofacitinib 10 mg BID compared to TNF inhibitors (aHR 1.60, 95% 1.09–2.36) and numerically greater in tofacitinib 5 mg BID compared to TNF inhibitors (aHR 1.35, 95% 0.91–2.01). Our findings might indeed justify, at least in part, the higher risk of fracture seen in this post-hoc analysis of the ORAL-surveillance trial. This is particularly true considering that disease activity reduction did not mediate the sclerostin increase and, therefore, this result appears to be independent from inflammation but merely “metabolic”. Notably, in the ORAL surveillance the disease activity was reduced similarly in both tofacitinib and TNF inhibitors groups, but the fracture incidence was significantly different. We might indeed speculate that the increased fracture risk seen in the ORAL surveillance might be attributed to the sclerostin increase. Interestingly, data from two large longitudinal studies showed that higher sclerostin levels were associated with coronary artery disease severity and greater mortality [23]. Further studies are needed to ascertain the precise role of sclerostin on vascular calcification and cardiovascular risk. Moreover current literature regarding the circulating sclerostin level and its role in RA patients are debatable [24, 25]. The lack of sclerostin or its antibody-mediated inhibition leads to an acceleration of RA-like disease in human TNFα transgenic mice with enhanced pannus formation and joint destruction [26].
Our study should be interpreted in light of strengths and limitations. We have collected detailed data on a homogeneous group of RA patients treated with tofacitinib, including the mSvdH score, BMD and fracture risk associated clinical factors. Moreover, we have analyzed a comprehensive panel of bone markers and modulators to investigate more precisely the effects on bone metabolism. However, our study is limited by the observational design and the small sample size, which was largely affected by the COVID-19 pandemic emergency and ORAL-surveillance results.
Conclusions
In conclusion, we observed a significant increase in sclerostin, a Wnt inhibitor, and a small but significant decrease in markers of bone formation, specifically B-ALP and P1NP. However no significant bone loss, both systemic and local, were observed during the year of treatment. Overall, our study sheds light on the complex interplay between tofacitinib, the Wnt system, and bone remodeling in RA patients. These findings contribute to a deeper understanding of the effects of JAK inhibitors on bone metabolism.
Data availability
Data of the analysis are available upon reasonable request.
Abbreviations
- RA:
-
Rheumatoid arthritis
- BMD:
-
Bone Mineral Density
- DMARDs:
-
Disease Modifying Anti-Rheumatic Drugs
- SvdH:
-
Sharp van der Heijde score
- BHI:
-
Bone Health Index
- MCI:
-
Metacarpal Index
- Dkk1:
-
Dickkopf-1
- iRMDs:
-
inflammatory rheumatic musculoskeletal diseases
- JAK:
-
Janus kinase
- TNF:
-
tumor necrosis factor
- CTX:
-
C-terminal telopeptide of type I collagen
- P1NP:
-
Procollagen I Intact N-Terminal Peptide
- B-ALP:
-
Bone Alkaline Phosphatase
- 25OHVitD:
-
25OH-Vitamin D
- PTH:
-
Parathyroid hormone
- OPG:
-
Osteoprotegerin
- RANKL:
-
Receptor activator of nuclear factor kappa-Β ligand
- SERM:
-
selective estrogen, receptor modulators
References
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19.
Adami G, Saag KG. Osteoporosis pathophysiology, epidemiology, and screening in rheumatoid arthritis. Curr Rheumatol Rep. 2019;21:34.
Adami G, Fassio A, Rossini M, Benini C, Pistillo F, Viapiana O et al. Bone loss occurs in Inflammatory Rheumatic Musculoskeletal diseases (iRMD) patients treated with low dose glucocorticoids, but is prevented by anti-osteoporosis medications. Arthritis Rheumatol 2023.
Adami G. Regulation of bone mass in inflammatory diseases. Best Pract Res Clin Endocrinol Metab 2021:101611.
Adami G, Fassio A, Rossini M, Caimmi C, Giollo A, Orsolini G et al. Osteoporosis in Rheumatic diseases. Int J Mol Sci 2019;20.
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, et al. Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007;13:156–63.
Rossini M, Gatti D, Adami S. Involvement of WNT/β-catenin signaling in the treatment of osteoporosis. Calcif Tissue Int. 2013;93:121–32.
Rossini M, Viapiana O, Adami S, Fracassi E, Idolazzi L, Dartizio C, et al. In patients with rheumatoid arthritis, Dickkopf-1 serum levels are correlated with parathyroid hormone, bone erosions and bone mineral density. Clin Exp Rheumatol. 2015;33:77–83.
Fassio A, Idolazzi L, Viapiana O, Benini C, Vantaggiato E, Bertoldo F, et al. In psoriatic arthritis Dkk-1 and PTH are lower than in rheumatoid arthritis and healthy controls. Clin Rheumatol. 2017;36:2377–81.
Adami G, Orsolini G, Adami S, Viapiana O, Idolazzi L, Gatti D, et al. Effects of TNF inhibitors on parathyroid hormone and wnt signaling antagonists in rheumatoid arthritis. Calcif Tissue Int. 2016;99:360–4.
Soós B, Szentpétery Á, Raterman HG, Lems WF, Bhattoa HP, Szekanecz Z. Effects of targeted therapies on bone in rheumatic and musculoskeletal diseases. Nat Rev Rheumatol. 2022;18:249–57.
van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–19.
Hansen KE, Mortezavi M, Nagy E, Wang C, Connell CA, Radi Z, et al. Fracture in clinical studies of tofacitinib in rheumatoid arthritis. Ther Adv Musculoskelet Dis. 2022;14:1759720X221142346.
Adami G, Orsolini G, Rossini M, Pedrollo E, Fratucello A, Fassio A et al. Changes in bone turnover markers and bone modulators during abatacept treatment. Scientific Reports 2023;13. https://www.scopus.com/inward/record.uri?eid=2-s2.0-85173669961&doi=10.1038%2fs41598-023-44374-2&partnerID=40&md5=92716e65ca73181c17efa08e6ea3c2b3
Adami G, Saag KG. Glucocorticoid-induced osteoporosis: 2019 concise clinical review. Osteoporosis International 2019. https://doi.org/10.1007%2Fs00198-019-04906-x
Hamar A, Szekanecz Z, Pusztai A, Czókolyová M, Végh E, Pethő Z, et al. Effects of one-year tofacitinib therapy on bone metabolism in rheumatoid arthritis. Osteoporos Int. 2021;32:1621–9.
Jorde R, Stunes AK, Kubiak J, Joakimsen R, Grimnes G, Thorsby PM, et al. Effects of vitamin D supplementation on bone turnover markers and other bone-related substances in subjects with vitamin D deficiency. Bone. 2019;124:7–13.
Rossini M, Maddali Bongi S, La Montagna G, Minisola G, Malavolta N, Bernini L, et al. Vitamin D deficiency in rheumatoid arthritis: prevalence, determinants and associations with disease activity and disability. Arthritis Res Ther. 2010;12:R216.
Adami G, Rossini M, Bogliolo L, Cantatore FP, Varenna M, Malavolta N, et al. An exploratory study on the role of vitamin D supplementation in improving pain and disease activity in rheumatoid arthritis. Mod Rheumatol. 2019;29:1059–62.
Vasikaran S, Cooper C, Eastell R, Griesmacher A, Morris HA, Trenti T, et al. International Osteoporosis Foundation and International Federation of Clinical Chemistry and Laboratory Medicine position on bone marker standards in osteoporosis. Clin Chem Lab Med. 2011;49:1271–4.
Orsolini G, Bertoldi I, Rossini M. Osteoimmunology in rheumatoid and psoriatic arthritis: potential effects of tofacitinib on bone involvement. Clin Rheumatol. 2020;39:727–36.
Adam S, Simon N, Steffen U, Andes FT, Scholtysek C, Müller DIH, et al. JAK inhibition increases bone mass in steady-state conditions and ameliorates pathological bone loss by stimulating osteoblast function. Sci Transl Med. 2020;12:eaay4447.
Frysz M, Gergei I, Scharnagl H, Smith GD, Zheng J, Lawlor DA, et al. Circulating sclerostin levels are positively related to coronary artery Disease Severity and related risk factors. J Bone Min Res. 2022;37:273–84.
Mao Y-M, Liao T, Ye Q-L, Wu G-C, Zhang Q, Tao S-S, et al. Increased circulating sclerostin levels in rheumatoid arthritis patients: an updated meta-analysis. Z Rheumatol. 2023;82:51–8.
Wehmeyer C, Stratis A, Pap T, Dankbar B. The role of the WNT inhibitor sclerostin in rheumatoid arthritis. Ann Rheum Dis. 2010;69:A21–21.
Wehmeyer C, Frank S, Beckmann D, Böttcher M, Cromme C, König U, et al. Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction. Sci Transl Med. 2016;8:330ra35.
Acknowledgements
Not applicable.
Funding
Research was supported by an unrestricted grant by Pfizer.
Author information
Authors and Affiliations
Contributions
Conceptualization, GO, MR; Data curation, GA, GO, MR; Formal analysis, GA; Investigation, GA, GO, MR, AF, AF, OV, EF, RB, DG; Project administration, MR; Supervision, MR; Validation, GA, GO, MR; Writing – original draft, GA; Writing – review & editing, GA, GO, MR, AF, AF, OV, EF, RB, DG.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the University of Verona ethic committee (prot. WI232128), in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was collected for each participant. Clinical trial number: not applicable.
Consent for publication
Not applicable.
Competing interests
Giovanni Adami has received advisory board honoraria, consultancy fees and/or speaker fees from Theramex, UCB, Lilly, Galapagos, Fresenius Kabi, Amgen, BMS, Abiogen and Pfizer. Davide Gatti has received advisory board honoraria, consultancy fees and/or speaker fees from Abiogen, Celgene, Eli-Lilly, Neopharmed-Gentili, Pfizer, UCB. Maurizio Rossini reports advisory board honoraria, consultancy fees and/or speaker fees from AbbVie, Amgen, BMS, Eli-Lilly, Galapagos, Menarini, Sandoz, Theramex, UCB, outside the submitted work. Ombretta Viapiana has received advisory board honoraria and speaker fees from Gilead, Fresenius Kabi, Biogen, Eli-Lilly, UCB, Abbvie, MSD, BMS. Angelo Fassio reports personal fees from Abiogen, Novartis, Neopharmed. The other authors have disclosed no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Adami, G., Orsolini, G., Rossini, M. et al. Effects of tofacitinib on bone turnover markers and bone modulators in patients with rheumatoid arthritis. BMC Rheumatol 8, 40 (2024). https://doi.org/10.1186/s41927-024-00414-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41927-024-00414-6